
LigGPT: Molecular Generation using a Transformer-Decoder Model

U. Deva Priyakumar, Viraj Bagal ,Rishal Aggarwal ,P. K. Vinod
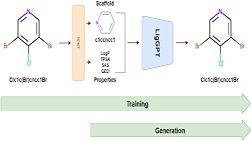
Application of deep learning techniques for the de novo generation of molecules, termed as inverse molecular design, has been gaining enormous traction in drug design. The representation of molecules in SMILES notation as a string of characters enables the usage of state of the art models in Natural Language Processing, such as the Transformers, for molecular design in general. Inspired by Generative Pre-Training (GPT) model that have been shown to be successful in generating meaningful text, we train a Transformer-Decoder on the next token prediction task using masked self-attention for the generation of druglike molecules in this study.
Keep Reading...Enhanced Sampling of Chemical Space for High Throughput Screening Applications using Machine Learning
Sarvesh Mehta ,Siddhartha Laghuvarapu ,Yashaswi Pathak ,Aaftaab Sethi ,Mallika Alvala ,U. Deva Priyakumar
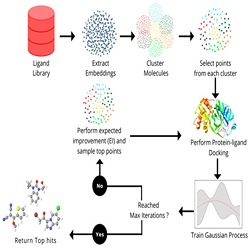
In drug discovery applications, high throughput virtual screening exercises are routinely performed to determine an initial set of candidate molecules referred to as "hits". In such an experiment, each molecule from large small-molecule drug library is evaluated for physical property such as the binding affinity (docking score) against a target receptor. In real-life drug discovery experiments, the drug libraries are extremely large but still a minor representation of the essentially infinite chemical space , and evaluation of physical property for each molecule in the library is not computationally feasible.
Keep Reading...
Modified Variable Kernel Length ResNets for Heart Murmur Detection and
Clinical Outcome Prediction using Multi-positional Phonocardiogram
Recording
U Deva Priyakumar, Vijay Vignesh Venkataramani , Akshit Garg
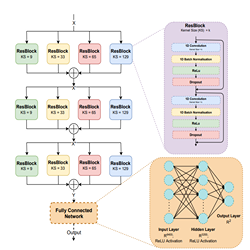
In our approach, the PCG recordings are first downsampled to 1000 Hz before being passed through a Butterworth’s low and high pass filter to remove baselinewanders and high-frequency noise present in the recordings. The PCG recordings are then broken down into10-second segments and normalized to bring all trainable samples to the same size.
Keep Reading...Carbodicarbenes and Striking Redox Transitions of their Conjugate Acids: Influence of NHC versus CAAC as Donor Substituents
Prof. U. Deva Priyakumar, Dr. Ramapada Dolai, Rahul Kumar, Dr. Benedict J. Elvers, Pradeep Kumar Pal, Dr. Benson Joseph, Dr. Rina Sikari, Mithilesh Kumar Nayak, Dr. Avijit Maiti, Dr. Tejender Singh, Nicolas Chrysochos, Dr. Arumugam Jayaraman, Dr. Ivo Krummenacher, Prof. Jagannath Mondal
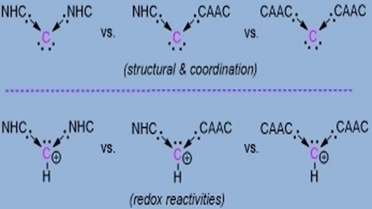
Herein, a new type of carbodicarbene (CDC) comprising two different classes of carbenes is reported; NHC and CAAC as donor substituents and compare the molecular structure and coordination to Au(I)Cl to those of NHC-only and CAAC-only analogues. The conjugate acids of these three CDCs exhibit notable redox properties. Their reactions with [NO][SbF6] were investigated. The reduction of the conjugate acid of CAAC-only based CDC with KC8 results in the formation of hydrogen abstracted/eliminated products, which proceed through a neutral radical intermediate, detected by EPR spectroscopy.
Keep Reading...Efficient and enhanced sampling of drug-like chemical space for virtual screening and molecular design using modern machine learning methods
Manan Goel, Rishal Aggarwal, Bhuvanesh Sridharan, Pradeep Kumar Pal, U. Deva Priyakumar
Drug design involves the process of identifying and designing novel molecules that have desirable properties and bind well to a given target receptor. Typically, such molecules are identified by screening large chemical libraries for desirable physicochemical properties and binding strength with the target protein. This traditional approach, however, has severe limitations as exhaustively screening every molecule in known chemical libraries is computationally infeasible.
Keep Reading...Tetra-Coordinated Boron-Functionalized Phenanthroimidazole-Based Zinc Salen as a Photocatalyst for the Cycloaddition of CO2 and Epoxides
U. Deva Priyakumar, Prakash Nayak, Anna Chandrasekar Murali, Pradeep Kumar Pal
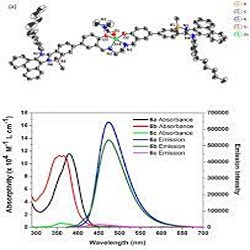
A unique B–N coordinated phenanthroimidazole-based zinc salen was synthesized. The zinc salen thus synthesized acts as a photocatalyst for the cycloaddition of carbon dioxide with terminal epoxides under ambient conditions. DFT study of the cycloaddition of carbon dioxide with terminal epoxide indicates the preference of the reaction pathway when photocatalyzed by zinc salen. We anticipate that this strategy will help to design new photocatalysts for CO2 fixation.
Keep Reading...MO-MEMES: A method for accelerating virtual screening using multi-objective Bayesian optimization
U. Deva Priyakumar, Sarvesh Mehta, Manan Goel
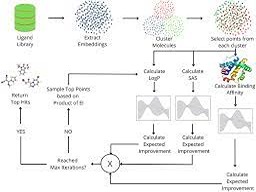
The pursuit of potential inhibitors for novel targets has become a very important problem especially over the last 2 years with the world in the midst of the COVID-19 pandemic. This entails performing high throughput screening exercises on drug libraries to identify potential “hits”. These hits are identified using analysis of their physical properties like binding affinity to the target receptor, octanol-water partition coefficient (LogP) and more. However, drug libraries can be extremely large and it is infeasible to calculate and analyze the physical properties for each of those molecules within acceptable time and moreover, each molecule must possess a multitude of properties apart from just the binding affinity.
Keep Reading...PLAS-5k: Dataset of Protein-Ligand Affinities from Molecular Dynamics for Machine Learning Applications
U. Deva Priyakumar, Divya B. Korlepara, C. S. Vasavi, Shruti Jeurkar, Pradeep Kumar Pal, Subhajit Roy, Sarvesh Mehta, Shubham Sharma, Vishal Kumar, Charuvaka Muvva, Bhuvanesh Sridharan, Akshit Garg, Rohit Modee, Agastya P. Bhati, Divya Nayar
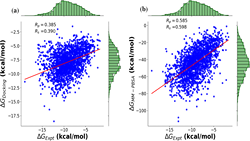
Computational methods and recently modern machine learning methods have played a key role in structure-based drug design. Though several benchmarking datasets are available for machine learning applications in virtual screening, accurate prediction of binding affinity for a protein-ligand complex remains a major challenge. New datasets that allow for the development of models for predicting binding affinities better than the state-of-the-art scoring functions are important. For the first time, we have developed a dataset, PLAS-5k comprised of 5000 protein-ligand complexes chosen from PDB database. The dataset consists of binding affinities along with energy components like electrostatic, van der Waals, polar and non-polar solvation energy calculated from molecular dynamics simulations using MMPBSA (Molecular Mechanics Poisson-Boltzmann Surface Area) method.
Keep Reading...COVID-19 Risk Stratification and Mortality Prediction in Hospitalized Indian Patients
P. K. Vinod, U. Deva Priyakumar
The clinical course of coronavirus disease 2019 (COVID-19) infection is highly variable with the vast majority recovering uneventfully but a small fraction progressing to severe disease and death. Appropriate and timely supportive care can reduce mortality and it is critical to evolve better patient risk stratification based on simple clinical data, so as to perform effective triage during strains on the healthcare infrastructure. This study presents risk stratification and mortality prediction models based on usual clinical data from 544 COVID-19 patients from New Delhi, India using machine learning methods. An XGboost classifier yielded the best performance on risk stratification (F1 score of 0.81). A logistic regression model yielded the best performance on mortality prediction (F1 score of 0.71).
Keep Reading...Development of predictive models of π-facial selectivity; a critical study of nucleophilic addition to sterically unbiased ketones
U.DevaPriyakumar, G.NarahariSastry, GoverdhanMehtab
Quantum chemical calculations at B3LYP/6-31G* and semiempirical levels have been performed on a series of sterically unbiased ketones, where facial differentiation during nucleophilic additions is electronically induced through distal functional groups. The face selectivity data for fifty-four substrates representing nine different skeleta were computed and compared with the available experimental data on thirty-eight of them. The predictive abilities of various computational methods such as, charge model, hydride model, LiH transition state model, Cieplak hyperconjugation effect estimated by NBO analysis and the cation complexation model have been evaluated.
Keep Reading...IMLE-Net: An Interpretable Multi-level Multi-channel Model for ECG Classification
U. Deva Priyakumar,Likith Reddy; Vivek Talwar; Shanmukh Alle; Raju. S. Bapi
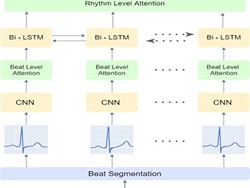
Early detection of cardiovascular diseases is crucial for effective treatment and an electrocardiogram (ECG) is pivotal for diagnosis. The accuracy of Deep Learning based methods for ECG signal classification has progressed in recent years to reach cardiologist-level performance. In clinical settings, a cardiologist makes a diagnosis based on the standard 12-channel ECG recording. Automatic analysis of ECG recordings from a multiple-channel perspective has not been given enough attention, so it is essential to analyze an ECG recording from a multiple-channel perspective.
Keep Reading...MolGPT: Molecular Generation Using a Transformer-Decoder Model
U. Deva Priyakumar,Viraj Bagal, Rishal Aggarwal, P. K. Vinod
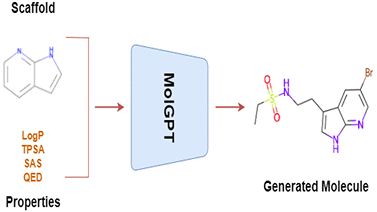
Application of deep learning techniques for de novo generation of molecules, termed as inverse molecular design, has been gaining enormous traction in drug design. The representation of molecules in SMILES notation as a string of characters enables the usage of state of the art models in natural language processing, such as Transformers, for molecular design in general. Inspired by generative pre-training (GPT) models that have been shown to be successful in generating meaningful text, we train a transformer-decoder on the next token prediction task using masked self-attention for the generation of druglike molecules in this study.
Keep Reading...The HIV-1 vpu transmembrane domain topology and formation of a hydrophobic interface with bst-2 are critical for vpu-mediated bst-2 downregulation
N Khan, S Padhi, P Patel, UD Priyakumar, S Jameel,

Viruses belonging to the M group of human immunodeficiency virus (HIV-1) are the most virulent among the four HIV-1 groups. One factor that distinguishes the M group HIV-1 from others is Vpu, a membrane localized accessory protein, which promotes the release of virions by neutralizing the antiviral host cell protein BST-2. To investigate if this activity is determined by the topology of Vpu or by conserved amino acid residues, we prepared chimeric forms of Vpu by replacing its transmembrane domain with those from its topological homologs. Although the chimeric Vpu proteins downregulated BST-2, these substantially reduced virus production as well. Molecular modeling studies on Vpu from different HIV-1 groups and the chimeric Vpu proteins showed that shape and the availability of a hydrophobic interface are more important for BST-2 antagonism than conservation of the amino acid sequence. Our data suggest that the HIV-1 Vpu-M protein has evolved topologically to interact with BST-2, and that the Vpu/BST-2 interface can be exploited as a target to limit HIV-1 replication.
Keep Reading...Structure-based drug repurposing: Traditional and advanced AI/ML-aided methods
Chinmayee Choudhury, N Arul Murugan, U Deva Priyakumar
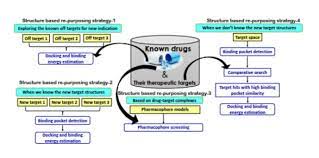
The current global health emergency in the form of the Coronavirus 2019 (COVID-19) pandemic has highlighted the need for fast, accurate, and efficient drug discovery pipelines. Traditional drug discovery projects relying on in vitro high-throughput screening (HTS) involve large investments and sophisticated experimental set-ups, affordable only to big biopharmaceutical companies. In this scenario, application of efficient state-of-the-art computational methods and modern artificial intelligence (AI)-based algorithms for rapid screening of repurposable chemical space [approved drugs and natural products (NPs) with proven pharmacokinetic profiles] to identify the initial leads is a powerful option to save resources and time. Structure-based drug repurposing is a popular in silico repurposing approach. In this review, we discuss traditional and modern AI-based computational methods and tools applied at various stages for structure-based drug discovery (SBDD) pipelines. Additionally, we highlight the role of generative models in generating molecules with scaffolds from repurposable chemical space.
Keep Reading...Benchmark study on deep neural network potentials for small organic molecules
R Modee, S Laghuvarapu, UD Priyakumar
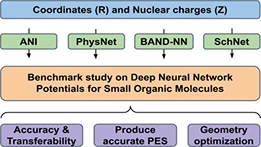
There has been tremendous advancement in machine learning (ML) applications in computational chemistry, particularly in neural network potentials (NNP). NNPs can approximate potential energy surface (PES) as a high dimensional function by learning from existing reference data, thereby circumventing the need to solve the electronic Schrödinger equation explicitly. As a result, ML accelerates chemical space exploration and property prediction compared to quantum mechanical methods. Novel ML methods have the potential to provide efficient means for predicting the properties of molecules. However, this potential has been limited by the lack of standard comparative evaluations. In this work, we compare four selected models, that is, ANI, PhysNet, SchNet, and BAND-NN, developed to represent the PES of small organic molecules. We evaluate these models for their accuracy and transferability on two different test sets (i) Small organic molecules of up to eight-heavy atoms on which ANI and SchNet achieve root mean square error (RMSE) of 0.55 and 0.60 kcal/mol, respectively. (ii) On random selection of molecules from the GDB-11 database with 10-heavy atoms, ANI achieves RMSE of 1.17 kcal/mol and SchNet achieves RMSE of 1.89 kcal/mol. We examine their ability to produce smooth meaningful surface by performing PES scans for bond stretch, angle bend, and dihedral rotations on...
Keep Reading...Spectra to Structure: Deep Reinforcement Learning for Molecular Inverse Problem
B Sridharan, S Mehta, Y Pathak, UD Priyakumar
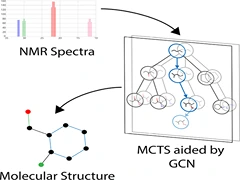
Spectroscopy is the study of how matter interacts with electromagnetic radiation. The spectra of any molecule are highly information-rich, yet the inverse relation of spectra to the corresponding molecular structure is still an unsolved problem. Nuclear magnetic resonance (NMR) spectroscopy is one such critical technique in the scientists’ toolkit to characterize molecules. In this work, a novel machine learning framework is proposed that attempts to solve this inverse problem by navigating the chemical space to find the correct structure given an NMR spectra.
Keep Reading...BiRDS – Binding Residue Detection from Protein Sequences using Deep ResNets
V Chelur, UD Priyakumar,
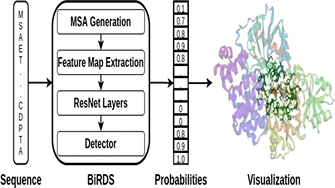
Protein–drug interactions play important roles in many biological processes and therapeutics. Predicting the binding sites of a protein helps to discover such interactions. New drugs can be designed to optimize these interactions, improving protein function. The tertiary structure of a protein decides the binding sites available to the drug molecule, but the determination of the 3D structure is slow and expensive. Conversely, the determination of the amino acid sequence is swift and economical. Although quick and accurate prediction of the binding site using just the sequence is challenging, the application of Deep Learning, which has been hugely successful in several biochemical tasks, makes it feasible.
Keep Reading...Benchmark study on deep neural network potentials for small organic molecules
R Modee, S Laghuvarapu, UD Priyakumar
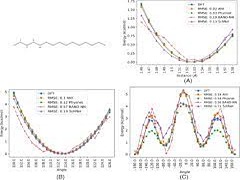
There has been tremendous advancement in machine learning (ML) applications in computational chemistry, particularly in neural network potentials (NNP). NNPs can approximate potential energy surface (PES) as a high dimensional function by learning from existing reference data, thereby circumventing the need to solve the electronic Schrödinger equation explicitly. As a result, ML accelerates chemical space exploration and property prediction compared to quantum mechanical methods. Novel ML methods have the potential to provide efficient means for predicting the properties of molecules. However, this potential has been limited by the lack of standard comparative evaluations. In this work, we compare four selected models, that is, ANI, PhysNet, SchNet, and BAND-NN, developed to represent the PES of small organic molecules. We evaluate these models for their accuracy and transferability on two different test sets
Keep Reading...Modern AI/ML Methods for Healthcare: Opportunities and Challenges
A Garg, VV Venkataramani, A Karthikeyan, U Priyakumar
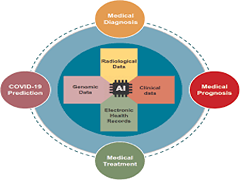
International Conference on Distributed Computing and Internet Technology Artificial Intelligence has seen a significant resurgence in the past decade in wide ranging technology and domain areas. Recent progress in digitisation and high influx of biomedical data have led to an unparalleled success of Machine Learning systems in healthcare, which is perceived to be a possible game changer for ‘healthcare to all’. This article gives an account of some of the current applications of AI solutions in the medical domains of diagnosis, prognosis and treatment.
Keep Reading...MolGPT: Molecular Generation Using a Transformer-Decoder Model
V Bagal, R Aggarwal, PK Vinod, UD Priyakumar
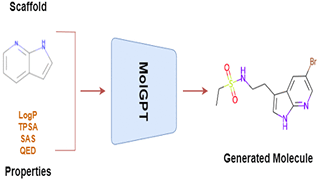
Application of deep learning techniques for de novo generation of molecules, termed as inverse molecular design, has been gaining enormous traction in drug design. The representation of molecules in SMILES notation as a string of characters enables the usage of state of the art models in natural language processing, such as Transformers, for molecular design in general. Inspired by generative pre-training (GPT) models that have been shown to be successful in generating meaningful text, we train a transformer-decoder on the next token prediction task using masked self-attention for the generation of druglike molecules in this study.
Keep Reading...Molecular Representations for Machine Learning Applications in Chemistry.
S Raghunathan, UD Priyakumar,
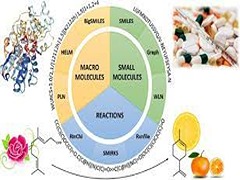
Machine learning (ML) methods enable computers to address problems by learning from existing data. Such applications are becoming commonplace in molecular sciences. Interest in applying ML techniques across chemical compound space, from predicting properties to designing molecules and materials is in the surge. Especially, ML models have started to accelerate computational chemistry, and are often as accurate as state-of-the-art electronic/atomistic models. Being an integral part of the ML architecture, representation of a molecular entity, uniquely encoded, plays a crucial role to what extent an ML model would be accurately predicting the desired property.
Keep Reading...Artificial Intelligence – Machine Learning for Chemical Sciences
A Karthikeyan, UD Priyakumar,
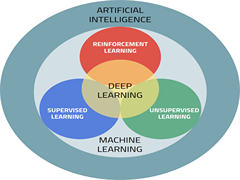
Research in molecular sciences witnessed the rise and fall of Artificial Intelligence (AI)/ Machine Learning (ML) methods, especially artificial neural networks, few decades ago. However, we see a major resurgence in the use of modern ML methods in scientific research during the last few years. These methods have had phenomenal success in the areas of computer vision, speech recognition, natural language processing (NLP), etc. This has inspired chemists and biologists to apply these algorithms to problems in natural sciences. Availability of high performance Graphics Processing Unit (GPU) accelerators, large datasets, new algorithms, and libraries has enabled this surge.
Keep Reading...DeepPocket: Ligand Binding Site Detection and Segmentation using 3D Convolutional Neural Networks.
R Aggarwal, A Gupta, V Chelur, CV Jawahar, UD Priyakumar,
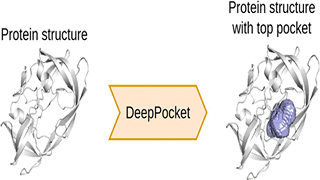
A structure-based drug design pipeline involves the development of potential drug molecules or ligands that form stable complexes with a given receptor at its binding site. A prerequisite to this is finding druggable and functionally relevant binding sites on the 3D structure of the protein. Although several methods for detecting binding sites have been developed beforehand, a majority of them surprisingly fail in the identification and ranking of binding sites accurately.
Keep Reading...Staufen-2 functions as a co-factor for enhanced nucleocytoplasmic trafficking of Rev-meditated viral RNA via the CRM1 pathway by making a tripartite complex.
K Balakrishnan, P Munusami, K Mohareer, UD Priyakumar, A Banerjee, T Luedde, S C Mande, C Munk, S Banerjee,
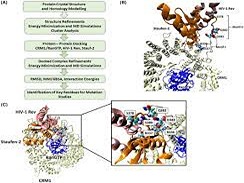
Nucleocytoplasmic shuttling of viral elements, supported by several host factors, is essential for the replication of the human immunodeficiency virus (HIV). HIV-1 uses a nuclear RNA export pathway mediated by viral protein Rev to transport its Rev response element (RRE)-containing partially spliced and unspliced transcripts aided by the host nuclear RNA export protein CRM1. The factor(s) interacting with the CRM1-Rev complex are potential antiretroviral target(s) and could serve as a retroviral model system to study nuclear export machinery adapted by these viruses. We earlier reported that cellular Staufen-2 interacts with Rev, facilitating viral-RNA export. Here, we identified the formation of a complex between Staufen-2, CRM1 and Rev. Molecular docking and simulations mapped the interacting residues in the RNA-binding Domain 4 of Staufen-2 as R336 and R337, which were experimentally verified to be critical for interactions among Staufen-2, CRM1 and Rev by mutational analysis.
Keep Reading...A Model of Graph Transactional Coverage Patterns with Applications to Drug Discovery
AS Reddy, PK Reddy, A Mondal, UD Priyakumar
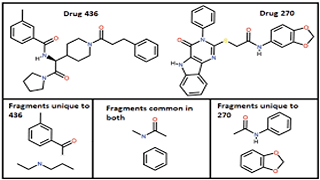
2021 IEEE 28th International Conference on High Performance Computing, SUBMITTED. Facilitating the discovery of drugs by combining diverse compounds is becoming prevalent, especially for treating complex diseases like cancers and HIV. A drug is a chemical compound structure and any sub-structure of a chemical compound is designated as a fragment. A chemical compound or a fragment can be modeled as a graph structure. Given a set of chemical compounds and their corresponding large set of fragments modeled as graph structures, we address the problem of identifying potential combinations of diverse chemical compounds, which cover a certain percentage of the set of fragments.
Keep Reading...MoleGuLAR: Molecule Generation Using Reinforcement Learning with Alternating Rewards
M Goel, S Raghunathan, S Laghuvarapu, UD Priyakumar
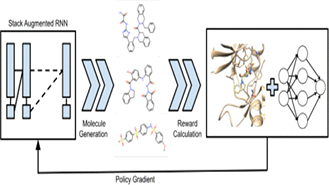
The design of new inhibitors for novel targets is a very important problem especially in the current scenario with the world being plagued by COVID-19. Conventional approaches such as high-throughput virtual screening require extensive combing through existing data sets in the hope of finding possible matches. In this study, we propose a computational strategy for de novo generation of molecules with high binding affinities to the specified target and other desirable properties for druglike molecules using reinforcement learning.
Keep Reading...Stereomutation in Tetracoordinate Centers via Stabilization of Planar Tetracoordinated Systems
K Yadav, U Lourderaj, UD Priyakumar
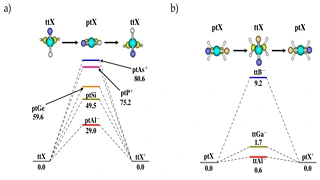
The quest for stabilizing planar forms of tetracoordinate carbon started five decades ago and intends to achieve interconversion between [R]- and [S]-stereoisomers without breaking covalent bonds. Several strategies are successful in making the planar tetracoordinate form a minimum on its potential energy surface. However, the first examples of systems where stereomutation is possible were reported only recently. In this study, the possibility of neutral and dications of simple hydrocarbons (cyclopentane, cyclopentene, spiropentane, and spiropentadiene) and their counterparts with the central carbon atom replaced by elements from groups 13, 14, and 15 are explored using ab initio MP2 calculations. The energy difference between the tetrahedral and planar forms decreases from row II to row III or IV substituents. Additionally, aromaticity involving the delocalization of the lone pair on the central atom appears to help in further stabilizing the planar form compared to the tetrahedral form, especially for the row II substituents.
Keep Reading...Synthesis and reactivity of NHC-coordinated phosphinidene oxide.
D Dhara, PK Pal, R Dolai, N Chrysochos, H Rawat, BJ Elvers, I Krummenacher, H Braunschweig, C Schulzke, V Chandrasekhar, UD Priyakumar, A Jana,D Dhara, PK Pal, R Dolai, N Chrysochos, H Rawat, BJ Elvers, I Krummenacher, H Braunschweig, C Schulzke, V Chandrasekhar, UD Priyakumar, A Jana,
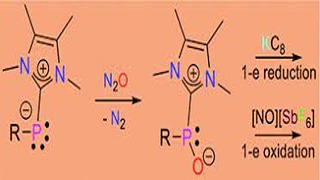
Here we report the synthesis of an N-heterocyclic carbene (NHC)-stabilised phosphinidene oxide by the controlled oxygenation of a phosphinidene under ambient conditions. This compound can be further oxygenated to a phosphinidene dioxide. The stoichiometric reduction of a phosphinidene oxide with KC8 resembles the pinacol coupling reaction–the reduction of a carbonyl compound. We also looked at the stoichiometric oxidation of NHC-coordinated phosphinidene, phosphinidene oxide and phosphinidene dioxide with [NO][SbF6].
Keep Reading...Desolvation of peptide bond by O to S substitution impacts protein stability .
B Khatri, S Ragunathan, S Chakraborti, Rahisuddin, S Kumaran, R Tadala, P Wagh, UD Priyakumar, J Chatterjee,
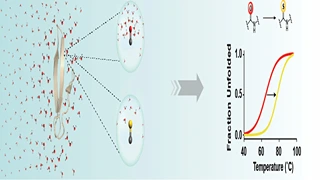
A C=O to C=S substitution in the amide bond dramatically alters the water structure around the thioamide bond, ultimately reducing the microenvironment polarity. The increased hydrophobicity of the modified peptide bond is utilized to amplify the thermostability of proteins by this single atom substitution.
Keep Reading...Mining Subgraph Coverage Patterns from Graph transactions.
AS Reddy, PK Reddy, A Mondal, UD Priyakumar,
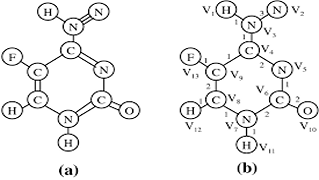
Pattern mining from graph transactional data (GTD) is an active area of research with applications in the domains of bioinformatics, chemical informatics and social networks. Existing works address the problem of mining frequent subgraphs from GTD. However, the knowledge concerning the coverage aspect of a set of subgraphs is also valuable for improving the performance of several applications. In this regard, we introduce the notion of subgraph coverage patterns (SCPs). Given a GTD, a subgraph coverage pattern is a set of subgraphs subject to relative frequency, coverage and overlap constraints provided by the user. We propose the Subgraph ID-based Flat Transactional (SIFT) framework for the efficient extraction of SCPs from a given GTD.
Keep Reading...SCONES: Self Consistent Neural Network for Protein Stability Prediction Upon Mutation.
Y Samaga BL., S Raghunathan, UD Priyakumar,
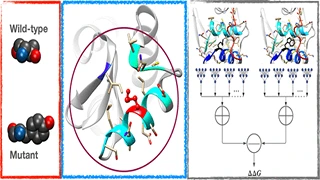
Engineering proteins to have desired properties by mutating amino acids at specific sites is commonplace. Such engineered proteins must be stable to function. Experimental methods used to determine stability at throughputs required to scan the protein sequence space thoroughly are laborious. To this end, many machine learning based methods have been developed to predict thermodynamic stability changes upon mutation. These methods have been evaluated for symmetric consistency by testing with hypothetical reverse mutations. In this work, we propose transitive data augmentation, evaluating transitive consistency with our new Stransitive data set, and a new machine learning based method, the first of its kind, that incorporates both symmetric and transitive properties into the architecture. Our method, called SCONES, is an interpretable neural network that predicts small relative protein stability changes for missense mutations that do not significantly alter the structure.
Keep Reading...DART – Deep Learning Enabled Topological Interaction Model for Energy Prediction of Metal Clusters and its Application in Identifying Unique Low Energy Isomers
R Modee, A Verma, K Joshi, UD Priyakumar,
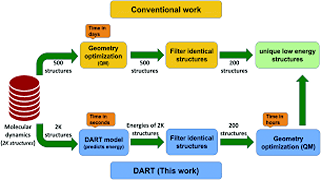
Recently, machine learning (ML) has proven to yield fast and accurate predictions of chemical properties to accelerate the discovery of novel molecules and materials. The majority of the work is on organic molecules, and much more work needs to be done for inorganic molecules, especially clusters. In the present work, we introduce a simple topological atomic descriptor called TAD, which encodes chemical environment information of each atom in the cluster. TAD is a simple and interpretable descriptor where each value represents the atom count in three shells. We also introduce the DART deep learning enabled topological interaction model, which uses TAD as a feature vector to predict energies of metal clusters, in our case gallium clusters with sizes ranging from 31 to 70 atoms. The DART model is designed based on the principle that the energy is a function of atomic interactions and allows us to model these complex atomic interactions to predict the energy. We further introduce a new dataset called GNC_31–70, which comprises structures and DFT optimized energies of gallium clusters with sizes ranging from 31 to 70 atoms. We show how DART can be used to accelerate the process of identification of low energy structures without geometry optimization.
Keep Reading...Clinico-Genomic analysis reveals mutations associated with COVID-19 disease severity: possible modulation by RNA structure.
P Metha, S Alle, A Chaturvedi, S Aparna, S Saifi, R Maurya, P Chattopadhay, P Devi, R Chauhan, A Kanakan, JS Vasudevan, R Sethuraman, C Subramanian, M Srivastava, A Chakravarthi, J Jacob, M Namagiri, V Konala, S Jha, UD Priyakumar, PK Vinod, R Pandey,
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) manifests a broad spectrum of clinical presentations, varying in severity from asymptomatic to mortality. As the viral infection spread, it evolved and developed into many variants of concern. Understanding the impact of mutations in the SARS-CoV-2 genome on the clinical phenotype and associated co-morbidities is important for treatment and preventionas the pandemic progresses. Based on the mild, moderate, and severe clinical phenotypes, we analyzed the possible association between both, the clinical sub-phenotypes and genomic mutations with respect to the severity and outcome of the patients.
Keep Reading...Host metabolic reprogramming in response to SARS-CoV-2 infection: A systems biology approach
STR Moolamalla, R Balasubramanian, R Chauhan, UD Priyakumar, PK Vinod,
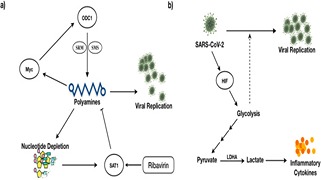
Understanding the pathogenesis of SARS-CoV-2 is essential for developing effective treatment strategies. Viruses hijack the host metabolism to redirect the resources for their replication and survival. The influence of SARS-CoV-2 on host metabolism is yet to be fully understood. In this study, we analyzed the transcriptomic data obtained from different human respiratory cell lines and patient samples (nasopharyngeal swab, peripheral blood mononuclear cells, lung biopsy, bronchoalveolar lavage fluid) to understand metabolic alterations in response to SARS-CoV-2 infection. We explored the expression pattern of metabolic genes in the comprehensive genome-scale network model of human metabolism, Recon3D, to extract key metabolic genes, pathways, and reporter metabolites under each SARS-CoV-2-infected condition.
Keep Reading...Linear Prediction Residual for Efficient Diagnosis of Parkinson’s Disease from Gait
S Alle, UD Priyakumar,
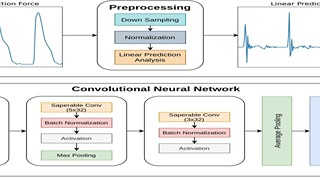
Parkinson’s Disease (PD) is a chronic and progressive neurological disorder that results in rigidity, tremors and postural instability. There is no definite medical test to diagnose PD and diagnosis is mostly a clinical exercise. Although guidelines exist, about 10–30% of the patients are wrongly diagnosed with PD. Hence, there is a need for an accurate, unbiased and fast method for diagnosis. In this study, we propose LPGNet, a fast and accurate method to diagnose PD from gait. LPGNet uses Linear Prediction Residuals (LPR) to extract discriminating patterns from gait recordings and then uses a 1D convolution neural network with depth-wise separable convolutions to perform diagnosis.
Keep Reading...Host metabolic reprogramming in response to SARS-Cov-2 infection,
STR Moolamalla, R Chauhan, UD Priyakumar, PK Vinod,
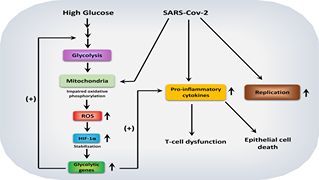
Understanding the pathogenesis of SARS-CoV-2 is essential for developing effective treatment strategies. Viruses hijack the host metabolism to redirect the resources for their replication and survival. The influence of SARS-CoV-2 on host metabolism is yet to be fully understood. In this study, we analyzed the transcriptomic data obtained from different human respiratory cell lines and patient samples (nasopharyngeal swab, peripheral blood mononuclear cells, lung biopsy, bronchoalveolar lavage fluid) to understand metabolic alterations in response to SARS-CoV-2 infection. We explored the expression pattern of metabolic genes in the comprehensive genome-scale network model of human metabolism, Recon3D, to extract key metabolic genes, pathways, and reporter metabolites under each SARS-CoV-2-infected condition. A SARS-CoV-2 core metabolic interactome was constructed for network-based drug repurposing. Our analysis revealed the host-dependent dysregulation of glycolysis, mitochondrial metabolism, amino acid metabolism, nucleotide metabolism, glutathione metabolism, polyamine synthesis, and lipid metabolism.
Keep Reading...MEMES: Machine learning framework for Enhanced MolEcular Screening
S Mehta, S Laghuvarapu, Y Pathak, A Sethi, M Alvala, UD Priyakumar,
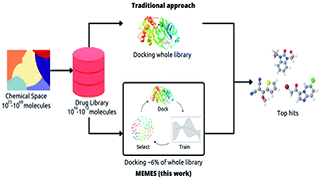
In drug discovery applications, high throughput virtual screening exercises are routinely performed to determine an initial set of candidate molecules referred to as “hits”. In such an experiment, each molecule from a large small-molecule drug library is evaluated in terms of physical properties such as the docking score against a target receptor. In real-life drug discovery experiments, drug libraries are extremely large but still there is only a minor representation of the essentially infinite chemical space, and evaluation of physical properties for each molecule in the library is not computationally feasible.
Keep Reading...IMLE-Net: An Interpretable Multi-level Multi-channel Model for ECG Classification.
L Reddy, V Talwar, S Alle, RS Bapi, UD Priyakumar,
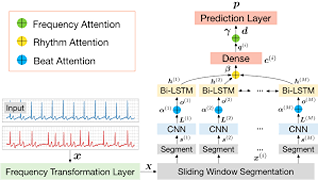
Early detection of cardiovascular diseases is crucial for effective treatment and an electrocardiogram (ECG) is pivotal for diagnosis. The accuracy of Deep Learning based methods for ECG signal classification has progressed in recent years to reach cardiologist-level performance. In clinical settings, a cardiologist makes a diagnosis based on the standard 12-channel ECG recording. Automatic analysis of ECG recordings from a multiple-channel perspective has not been given enough attention, so it is essential to analyze an ECG recording from a multiple-channel perspective.
Keep Reading...Multiscale Modeling of Wobble to Watson-Crick-like Guanine-Uracil Tautomerization Pathways in RNA
S Chandorkar, S Raghunathan, T Jaganade, UD Priyakumar,
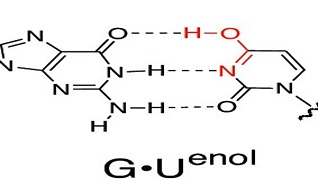
Energetically unfavorable Watson–Crick (WC)-like tautomeric forms of nucleobases are known to introduce spontaneous mutations, and contribute to replication, transcription, and translation errors. Recent NMR relaxation dispersion techniques were able to show that wobble (w) G•U mispair exists in equilibrium with the short-lived, low-population WC-like enolic tautomers. Presently, we have investigated the wG•U → WC-like enolic reaction pathway using various theoretical methods: quantum mechanics (QM), molecular dynamics (MD), and combined quantum mechanics/molecular mechanics (QM/MM). The previous studies on QM gas phase calculations were inconsistent with experimental data. We have also explored the environmental effects on the reaction energies by adding explicit water.
Keep Reading...Ion Selectivity and Permeation Mechanism in a Cyclodextrin-based Channel
P Musunuru, S Padhi, UD Priyakumar,
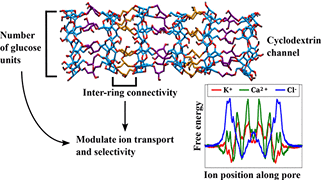
Synthetic ion channels are a promising technology in the medical and materials sciences because of their ability to conduct ions. Channels based on cyclodextrin, a cyclic oligomer of glucose, are of particular interest because of their nontoxicity and biocompatibility. Using molecular dynamics-based free energy calculations, this study identifies cyclodextrin channel types that are best suited to serve as synthetic ion channels. Free energy profiles show that the connectivity in the channel determines whether the channel is cation-selective or anion-selective.
Keep Reading...MMBERT: Multimodal BERT Pretraining for Improved Medical VQA,
Y Khare, V Bagal, M Mathew, A Devi, UD Priyakumar, CV Jawahar,
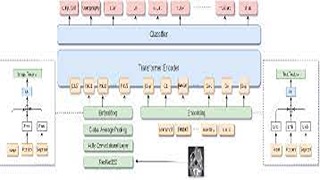
Images in the medical domain are fundamentally differentfrom the general domain images. Consequently, it is infeasible to directly employ general domain Visual Question Answering (VQA) models for the medical domain. Additionally,medical image annotation is a costly and time-consuming process. To overcome these limitations, we propose a solution inspired by self-supervised pretraining of Transformer-style architectures for NLP, Vision, and Language tasks. Our methodinvolves learning richer medical image and text semantic representations using Masked Vision-Language Modeling as thepretext task on a large medical image+caption dataset.
Keep Reading...Machine learning based clinical decision support system for early covid-19 mortality prediction
A Karthikeyan, A Garg, PK Vinod, UD Priyakumar,
The coronavirus disease 2019 (COVID-19), caused by the virus SARS-CoV-2, is an acute respiratory disease that has been classified as a pandemic by the World Health Organization (WHO). The sudden spike in the number of infections and high mortality rates have put immense pressure on the public healthcare systems. Hence, it is crucial to identify the key factors for mortality prediction to optimize patient treatment strategy. Different routine blood test results are widely available compared to other forms of data like X-rays, CT-scans, and ultrasounds for mortality prediction. This study proposes machine learning (ML) methods based on blood tests data to predict COVID-19 mortality risk. A powerful combination of five features: neutrophils, lymphocytes, lactate dehydrogenase (LDH), high-sensitivity C-reactive protein (hs-CRP), and age helps to predict mortality with 96% accuracy. Various ML models (neural networks, logistic regression, XGBoost, random forests, SVM, and decision trees) have been trained and performance compared to determine the model that achieves consistently high accuracy across the days that span the disease.
Keep Reading...Learning Atomic Interactions through Solvation Free Energy Prediction Using Graph Neural Networks
Y Pathak, S Mehta, UD Priyakumar,
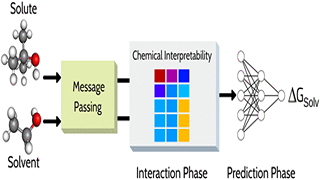
Solvation free energy is a fundamental property that influences various chemical and biological processes, such as reaction rates, protein folding, drug binding, and bioavailability of drugs. In this work, we present a deep learning method based on graph networks to accurately predict solvation free energies of small organic molecules. The proposed model, comprising three phases, namely, message passing, interaction, and prediction, is able to predict solvation free energies in any generic organic solvent with a mean absolute error of 0.16 kcal/mol.
Keep Reading...The HIV-1 vpu transmembrane domain topology and formation of a hydrophobic interface with bst-2 are critical for vpu-mediated bst-2 downregulation
N Khan, S Padhi, P Patel, UD Priyakumar, S Jameel,

Viruses belonging to the M group of human immunodeficiency virus (HIV-1) are the most virulent among the four HIV-1 groups. One factor that distinguishes the M group HIV-1 from others is Vpu, a membrane localized accessory protein, which promotes the release of virions by neutralizing the antiviral host cell protein BST-2. To investigate if this activity is determined by the topology of Vpu or by conserved amino acid residues, we prepared chimeric forms of Vpu by replacing its transmembrane domain with those from its topological homologs. Although the chimeric Vpu proteins downregulated BST-2, these substantially reduced virus production as well.
Keep Reading...Deep learning enabled inorganic material generator.
Y Pathak, KS Juneja, G Varma, M Ehara, UD Priyakumar,
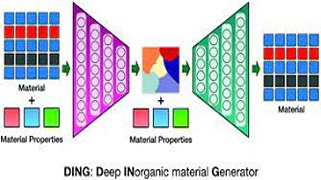
Recent years have witnessed utilization of modern machine learning approaches for predicting the properties of materials using available datasets. However, to identify potential candidates for material discovery, one has to systematically scan through a large chemical space and subsequently calculate the properties of all such samples. On the other hand, generative methods are capable of efficiently sampling the chemical space and can generate molecules/materials with desired properties.
Keep Reading...Machine learning for accurate force calculations in molecular dynamics simulations
P Pattnaik, S Raghunathan, T Kalluri, P Bhimalapuram, CV Jawahar, UD Priyakumar
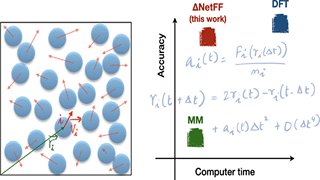
The computationally expensive nature of ab initio molecular dynamics simulations severely limits its ability to simulate large system sizes and long time scales, both of which are necessary to imitate experimental conditions. In this work, we explore an approach to make use of the data obtained using the quantum mechanical density functional theory (DFT) on small systems and use deep learning to subsequently simulate large systems by taking liquid argon as a test case. A suitable vector representation was chosen to represent the surrounding environment of each Ar atom, and a Δ-NetFF machine learning model, where the neural network was trained to predict the difference in resultant forces obtained by DFT and classical force fields, was introduced.
Keep Reading...Transition between [R]- and [S]-stereoisomers without bond breaking
S Raghunathan, K Yadav, VC Rojisha, T Jaganade, V Prathyusha, S Bikkina, U Lourderaj, UD Priyakumar.
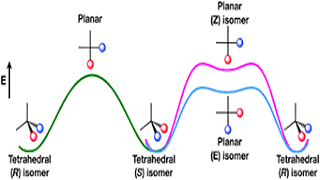
The fifty-year old proposal of a nondissociative racemization reaction of a tetracoordinated tetrahedral center from one enantiomer to another via a planar transition state by Hoffmann and coworkers has been explored by many research groups over the past five decades.
Keep Reading...Enantioseparation and chiral induction in Ag 29 nanoclusters with intrinsic chirality
H Yoshida, M Ehara, UD Priyakumar, T Kawai, T Nakashima,
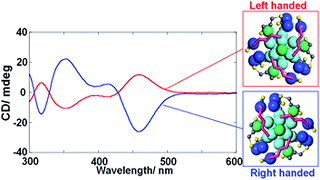
The optical activity of a metal nanocluster (NC) is induced either by an asymmetric arrangement of constituents or by a dissymmetric field of a chiral ligand layer. Herein, we unveil the origin of chirality in Ag29 NCs, which is attributed to the intrinsically chiral atomic arrangement.
Keep Reading...Urea-water solvation of protein side chain models
T Jaganade, A Chattopadhyay, S Raghunathan, UD Priyakumar.
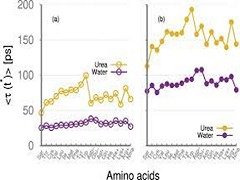
Aqueous urea stabilizes the unfolded states of protein due to their ability to solvate both hydrophilic and hydrophobic residues favorably. The nature of interactions that stabilize different types of amino acid side chains in their solvent exposed state is still not understood. To gain insights into the molecular level details of urea interactions with proteins in their unfolded states, we have performed atomistic molecular dynamics simulations and free energy calculations using the thermodynamic integration method on model systems representing side chains of all amino acids in different solvent environments (water and varying concentrations of aqueous urea). A systematic analysis of structural, energetic and dynamic parameters has been done to understand the detailed atomistic mechanism.
Keep Reading...BAND NN: A deep learning framework for energy prediction and geometry optimization of organic small molecules
S Laghuvarapu, Y Pathak, UD Priyakumar,
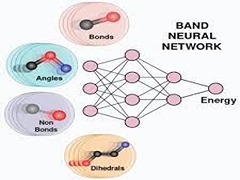
Recent advances in artificial intelligence along with the development of large data sets of energies calculated using quantum mechanical (QM)/density functional theory (DFT) methods have enabled prediction of accurate molecular energies at reasonably low computational cost. However, machine learning models that have been reported so far require the atomic positions obtained from geometry optimizations using high-level QM/DFT methods as input in order to predict the energies and do not allow for geometry optimization. In this study, a transferable and molecule size-independent machine learning model bonds (B), angles (A), nonbonded (N) interactions, and dihedrals (D) neural network (BAND NN) based on a chemically intuitive representation inspired by molecular mechanics force fields is presented.
Keep Reading...Chemically interpretable graph interaction network for prediction of pharmacokinetic properties of drug-like molecules
Y Pathak, S Laghuvarapu, S Mehta, U Priyakumar.
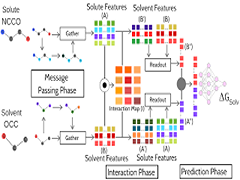
Solubility of drug molecules is related to pharmacokinetic properties such as absorption and distribution, which affects the amount of drug that is available in the body for its action. Computational or experimental evaluation of solvation free energies of drug-like molecules/solute that quantify solubilities is an arduous task and hence development of reliable computationally tractable models is sought after in drug discovery tasks in pharmaceutical industry. Here, we report a novel method based on graph neural network to predict solvation free energies. Previous studies considered only the solute for solvation free energy prediction and ignored the nature of the solvent, limiting their practical applicability.
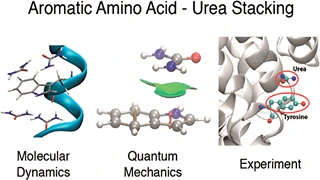
Keep Reading...Urea-aromatic interactions in biology
S Raghunathan, T Jaganade, UD Priyakumar,
Noncovalent interactions are key determinants in both chemical and biological processes. Among such processes, the hydrophobic interactions play an eminent role in folding of proteins, nucleic acids, formation of membranes, protein-ligand recognition, etc.. Though this interaction is mediated through the aqueous solvent, the stability of the above biomolecules can be highly sensitive to any small external perturbations, such as temperature, pressure, pH, or even cosolvent additives, like, urea-a highly soluble small organic molecule utilized by various living organisms to regulate osmotic pressure. A plethora of detailed studies exist covering both experimental and theoretical regimes, to understand how urea modulates the stability of biological macromolecules.
Keep Reading...Selectivity and transport in aquaporins from molecular simulation studies
S Padhi, UD Priyakumar,
The transport of water through aquaporins is a dynamic process that involves rapid movement of a chain of water molecules through the pore of the aquaporin. Structures of aquaporins solved using X-ray crystallography have provided some insights into how water is transported through these channels, and how certain structural features of the pore might help exclude other solutes from passing through the pore. However, such techniques provide only a static picture, and a dynamic picture of the transport and selectivity mechanism at work in aquaporins is possible with molecular dynamics (MD) simulations. In MD simulations, the forces between the different atoms in a system are computed, and the atoms are then allowed to move under the influence of these forces.
Keep Reading...Energetic, structural and dynamic properties of nucleobase-urea interactions that aid in urea assisted RNA unfolding
T Jaganade, A Chattopadhyay, NM Pazhayam, UD Priyakumar.
Understanding the structure-function relationships of RNA has become increasingly important given the realization of its functional role in various cellular processes. Chemical denaturation of RNA by urea has been shown to be beneficial in investigating RNA stability and folding. Elucidation of the mechanism of unfolding of RNA by urea is important for understanding the folding pathways. In addition to studying denaturation of RNA in aqueous urea, it is important to understand the nature and strength of interactions of the building blocks of RNA. In this study, a systematic examination of the structural features and energetic factors involving interactions between nucleobases and urea is presented.
Keep Reading...Cholic acid-derived amphiphile which combats gram-positive bacteria- mediated infections via disintegration of lipid clusters
S Kumar, J Thakur, K Yadav, S Padhi, UD Priyakumar, U Dasgupta, L Thukral, A Bajaj.
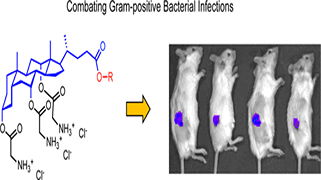
Inappropriate and uncontrolled use of antibiotics results in the emergence of antibiotic resistance, thereby threatening the present clinical regimens to treat infectious diseases. Therefore, new antimicrobial agents that can prevent bacteria from developing drug resistance are urgently needed. Selective disruption of bacterial membranes is the most effective strategy for combating microbial infections as accumulation of genetic mutations will not allow for the emergence of drug resistance against these antimicrobials. In this work, we tested cholic acid (CA) derived amphiphiles tethered with different alkyl chains for their ability to combat Gram-positive bacterial infections. In-depth biophysical and biomolecular simulation studies suggested that the amphiphile with a hexyl chain (6) executes more effective interactions with Gram-positive bacterial membranes as compared to other hydrophobic counterparts.
Keep Reading...Gold‐palladium nanocluster catalysts for homocoupling: electronic structure and interface dynamics
M Ehara, UD Priyakumar,
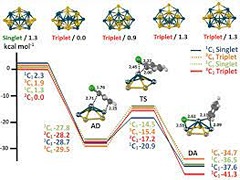
The gold-palladium (Au−Pd) bimetallic nanocluster (NC) catalyst in colloidal phase performs the homocoupling reaction of various aryl chlorides (Ar−Cl) under ambient conditions. We have systematically investigated various aspects of the Au−Pd NC catalysts with respect to this homocoupling reaction by using density functional theory (DFT) calculations, genetic algorithm (GA) approaches, and molecular dynamics (MD) simulations.
Keep Reading...omparative study of the efficiency of Au, Ag, Pd and Pt based mono and bimetallic trimer clusters for the CO oxidation reaction
S Gurtu, S Rai, UD Priyakumar,
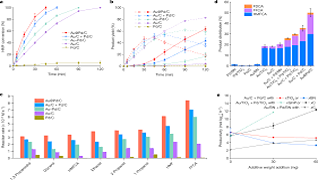
Comparative study of the efficiency of Au, Ag, Pd and Pt based mono and bimetallic trimer clusters for the CO oxidation reaction Saumya Gurtua, Sandhya Raib and U. Deva Priyakumara* aCenter for Computational Natural Sciences and Bioinformatics, International Institute of Information Technology, Hyderabad-500 032, India bTheoretical Science Unit, Jawaharlal Nehru Centre for Advanced Scientific Research, Jakkur, Bengaluru-560 001, India
Keep Reading...Computational modeling of the catalytic mechanism of hydroxymethylbilane synthase
N Bung, A Roy, UD Priyakumar, G Bulusu.
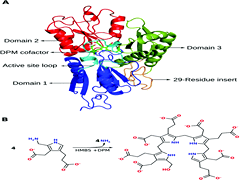
Hydroxymethylbilane synthase (HMBS), the third enzyme in the heme biosynthesis pathway, catalyzes the formation of 1-hydroxymethylbilane (HMB) by a stepwise polymerization of four molecules of porphobilinogen (PBG) using the dipyrromethane (DPM) cofactor. The mechanism by which HMBS polymerizes four units of PBG has not been elucidated to date. In vitro and in silico studies on HMBS have suggested certain residues with catalytic importance, but their specific role in the catalysis is unclear. To understand the catalytic mechanism of HMBS, quantum mechanical (QM) calculations were performed on model systems obtained from the active site of the human HMBS enzyme. The addition of one molecule of PBG to the DPM cofactor is carried out in four steps: (1) protonation of the substrate, PBG; (2) deamination of PBG; (3) electrophilic addition of the deaminated substrate to the terminal pyrrole ring of the enzyme-bound DPM cofactor and (4) deprotonation of the carbon atom at the α-position of the second ring of DPM. Based on the energy profiles from the QM calculations on cluster models, R26 is proposed to be the best suitable proton donor to the PBG moiety, which aids in the deamination of the substrate. During the electrophilic addition step, the intermediate formed is stabilized by the carboxylate side chain of the...
Keep Reading...Recent advancements in computing reliable binding free energies in drug discovery projects
NA Murugan, V Poongavanam, UD Priyakumar, In: C. Mohan (Eds.)
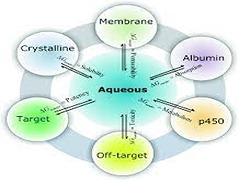
In recent times, our healthcare system is being challenged by many drug-resistant microorganisms and ageing-associated diseases for which we do not have any drugs or drugs with poor therapeutic profile. With pharmaceutical technological advancements, increasing computational power and growth of related biomedical fields, there have been dramatic increase in the number of drugs approved in general, but still way behind in drug discovery for certain class of diseases. Now, we have access to bigger genomics database, better biophysical methods, and knowledge about chemical space with which we should be able to easily explore and predict synthetically feasible compounds for the lead optimization process. In this chapter, we discuss the limitations and highlights of currently available computational methods used for protein–ligand binding affinities estimation and this includes force-field, ab initio electronic structure theory and machine learning approaches.
Keep Reading...Quantum mechanical investigation of the nature of nucleobase-urea stacking interaction, a crucial driving force in RNA unfolding in aqueous urea
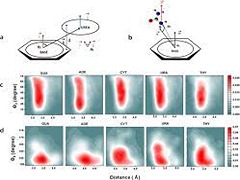
N Alodia, T Jaganade, UD Priyakumar,
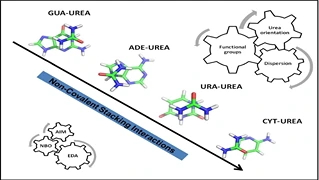
Urea-assisted denaturation of protein and RNA has been shown to be a valuable tool to study their stabilities and folding phenomena. It has been shown that stacking interactions between nucleobases and urea are one of the driving forces of denaturation. In this study, the ability of urea to form unconventional stacking interactions with RNA bases is investigated by performing high-level quantum calculations (RI-MP2/aug-cc-pVDZ level) on a few thousands of model systems. Four systems were considered based on the RNA nucleobases (GUA, ADE, CYT, and URA) for the investigation.
Keep Reading...A probabilistic framework for constructing temporal relations in replica exchange molecular trajectories
A Chattopadhyay, M Zheng, MP Waller, UD Priyakumar.
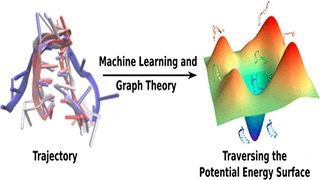
Knowledge of the structure and dynamics of biomolecules is essential for elucidating the underlying mechanisms of biological processes. Given the stochastic nature of many biological processes, like protein unfolding, it is almost impossible that two independent simulations will generate the exact same sequence of events, which makes direct analysis of simulations difficult. Statistical models like Markov chains, transition networks, etc. help in shedding some light on the mechanistic nature of such processes by predicting long-time dynamics of these systems from short simulations. However, such methods fall short in analyzing trajectories with partial or no temporal information, for example, replica exchange molecular dynamics or Monte Carlo simulations.
Keep Reading...Model molecules to classify CH⋯ O hydrogen-bonds, AM Vibhute
UD Priyakumar, A Ravi, KM Sureshan.
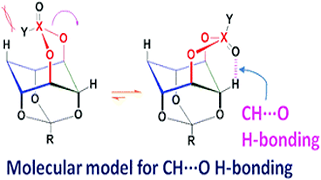
We developed a set of conformationally locked molecules each of which makes a single CH⋯O H-bond/short contact and has different electron density at the acceptor oxygen atom.
Keep Reading...pH-mediated gating and formate transport mechanism in the Escherichia coli formate channel
S Padhi, LK Reddy, UD Priyakumar,
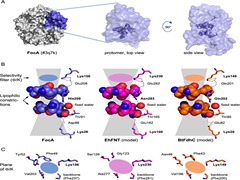
Formate channels play a crucial role in the metabolism of several different kinds of bacteria by bringing about uptake and export of formate ions. The current study investigates the structure, dynamics and ion channel activity of the formate channel FocA from Escherichia coli by employing extensive molecular dynamics (MD) simulations. The channel, known to be pH-sensitive, is modelled under different pH conditions by considering two different protonation states of a histidine residue. The results show that a fall in pH brings about an enhancement of the formate-conducting activity of the channel. The increased conductivity is a consequence of an overall widening of the pore at low pH, with the widening being brought about by movements of a pore-facing helical domain and a loop region.
Keep Reading...Role of urea–aromatic stacking interactions in stabilizing the aromatic residues of the protein in urea-induced denatured state
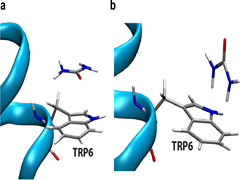
S Goyal, A Chattopadhyay, K Kasavajhala, UD Priyakumar,
A delicate balance of different types of intramolecular interactions makes the folded states of proteins marginally more stable than the unfolded states. Experiments use thermal, chemical, or mechanical stress to perturb the folding equilibrium for examining protein stability and the protein folding process. Elucidation of the mechanism by which chemical denaturants unfold proteins is crucial; this study explores the nature of urea–aromatic interactions relevant in urea-assisted protein denaturation. Free energy profiles corresponding to the unfolding of Trp-cage miniprotein in the presence and absence of urea at three different temperatures demonstrate the distortion of the hydrophobic core to be a crucial step. Exposure of the Trp6 residue to the solvent is found to be favored in the presence of urea.
Keep Reading...Temperature dependence of the stability of ion pair interactions, and its implications on the thermostability of proteins from thermophiles
S Bikkina, AP Bhati, S Padhi, UD Priyakumar,
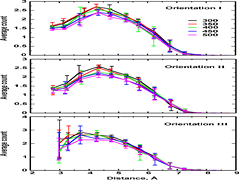
An understanding of the determinants of the thermal stability of thermostable proteins is expectedto enable design of enzymes that can be employed in industrial biocatalytic processes carried out at hightemperatures. A major factor that has been proposed to stabilize thermostable proteins is the high occurrenceof salt bridges. The current study employs free energy calculations to elucidate the thermodynamics of theformation of salt bridge interactions and the temperature dependence, using acetate and methylguanidium ionsas model systems. Three different orientations of the methylguanidinium approaching the carboxylate grouphave been considered for obtaining the free energy profiles.
Keep Reading...Modeling complex biomolecular systems and processes using molecular mechanics force fields and molecular dynamics simulations
S Padhi, UD Priyakumar, In: D. Kumar (Eds.)
Molecular dynamics (MD) simulations complement experimental structure determination methods like X-ray crystallography and NMR spectroscopy in elucidating macromolecular structure and behaviour. While these experimental techniques are capable of providing reliable structural information, understanding the atomistic details of structure-function relationships of biomolecules is limited. On the other hand, MD simulations are being extensively used to understand molecular processes, structure-function relationships, intramolecular/intermolecular interactions, and so on.
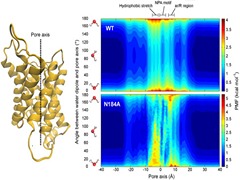
Keep Reading...Microsecond simulation of human aquaporin 2 reveals structural determinants of water permeability and selectivity
S Padhi, UD Priyakumar
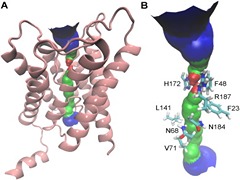
Human aquaporin 2 (AQP2) from the family of aquaporins assumes great physiological importance, owing to its association with nephrogenic diabetes insipidus (NDI). The present study provides detailed insights into the transport properties of AQP2 with the use of microsecond-scale molecular dynamics simulations, and explains how these channels conduct water molecules while at the same time excluding other molecules. Water transport is seen to be diffusion-limited, with a barrier of only 1.6 kcal mol− 1, and the channel is more water-permeable than other known aquaporins. A constriction site with a pore-facing phenylalanine and arginine is proposed to serve as a selectivity filter as well as a gate modulating the conductance state of the channel.
Keep Reading...Urea mimics nucleobases by preserving the helical integrity of B-DNA duplexes via hydrogen bonding and stacking interactions
G Suresh, S Padhi, I Patil, UD Priyakumar
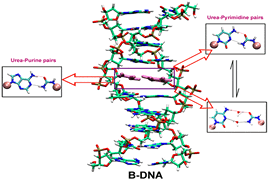
Urea lesions are formed in DNA because of free radical damage of the thymine base, and their occurrence in DNA blocks DNA polymerases, which has deleterious consequences. Recently, it has been shown that urea is capable of forming hydrogen bonding and stacking interactions with nucleobases, which are responsible for the unfolding of RNA in aqueous urea. Base pairing and stacking are inherent properties of nucleobases; because urea is able to form both, this study attempts to investigate if urea can mimic nucleobases in the context of nucleic acid structures by examining the effect of introducing urea lesions complementary to the four different nucleobases on the overall helical integrity of B-DNA duplexes and their thermodynamic stabilities using molecular dynamics (MD) simulations. The MD simulations resulted in stable duplexes without significant changes in the global B-DNA conformation.
Keep Reading...Urea–aromatic stacking and concerted urea transport: conserved mechanisms in urea transporters revealed by molecular dynamics
S Padhi, UD Priyakumar,
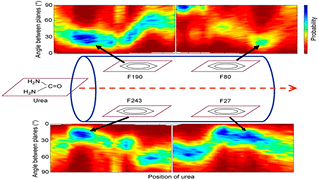
Urea transporters are membrane proteins that selectively allow urea molecules to pass through. It is not clear how these transporters allow rapid conduction of urea, a polar molecule, in spite of the presence of a hydrophobic constriction lined by aromatic rings. The current study elucidates the mechanism that is responsible for this rapid conduction by performing free energy calculations on the transporter dvUT with a cumulative sampling time of about 1.3 μs.
Keep Reading...Ligand-induced stabilization of a duplex-like architecture is crucial for the switching mechanism of the SAM-III riboswitch
G Suresh, H Srinivasan, S Nanda, UD Priyakumar,
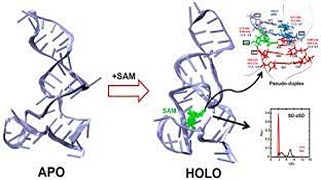
Riboswitches are structured RNA motifs that control gene expression by sensing the concentrations of specific metabolites and make up a promising new class of antibiotic targets. S-Adenosylmethionine (SAM)-III riboswitch, mainly found in lactic acid bacteria, is involved in regulating methionine and SAM biosynthetic pathways. SAM-III riboswitch regulates the gene expression by switching the translation process on and off with respect to the absence and presence of the SAM ligand, respectively. In this study, an attempt is made to understand the key conformational transitions involved in ligand binding using atomistic molecular dynamics (MD) simulations performed in an explicit solvent environment. G26 is found to recognize the SAM ligand by forming hydrogen bonds, whereas the absence of the ligand leads to opening of the binding pocket.
Keep Reading...Structure, interaction, and dynamics of Au/Pd bimetallic nanoalloys dispersed in aqueous ethylpyrrolidone, a monomeric moiety of polyvinylpyrrolidone
A Gupta, B Boekfa, H Sakurai, M Ehara, UD Priyakumar,
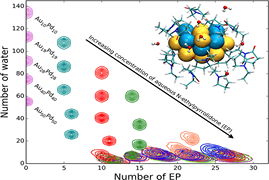
Bimetallic nanoparticles (NPs) have been shown to exhibit certain advantages over pure NPs in catalysis due to a synergistic effect. It is common to disperse NPs in a polymer matrix such as polyvinylpyrrolidone (PVP) to prevent flocculation, which imparts considerable electronic effects on the NPs. In the present study, the interactions between aqueous solutions of N-ethylpyrrolidone (EP, system chosen to model the monomeric form of PVP) and Au/Pd bimetallic NPs, which are relevant in catalysis, have been investigated using molecular dynamics simulations and density functional theory (DFT) method. The adequacy of the force fields used was assessed based on their ability to reproduce the structures and adsorption energies obtained using DFT calculations.
Keep Reading...Cooperation of hydrophobic gating, knock-on effect, and ion binding determines ion selectivity in the p7 channel
S Padhi, UD Priyakumar
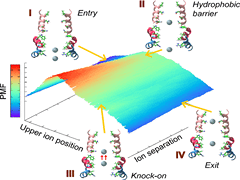
Ion channels selectively allow certain ions to pass through at much higher rates than others, and thereby modulate ionic concentrations across cell membranes. The current molecular dynamics study elucidates the intricate mechanisms that render ion selectivity to the viral channel p7 by employing free energy calculations. Free energy barriers of 5.4 and 19.4 kcal mol–1 for K+ and Ca2+, respectively, explain the selectivity of the channel reported in experiments. Initially, the permeating ions encounter a hydrophobic barrier followed by stabilization in an ion-binding site.
Keep Reading...Dynamic ligand-based pharmacophore modeling and virtual screening to identify mycobacterial cyclopropane synthase inhibitors
C Choudhury, UD Priyakumar, GN Sastry,
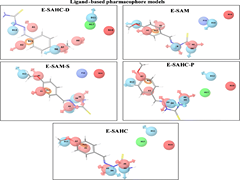
Multidrug resistance in Mycobacterium tuberculosis (M. Tb) and its coexistence with HIV are the biggest therapeutic challenges in anti-M. Tb drug discovery. The current study reports a Virtual Screening (VS) strategy to identify potential inhibitors of Mycobacterial cyclopropane synthase (CmaA1), an important M. Tb target considering the above challenges. Five ligand-based pharmacophore models were generated from 40 different conformations of the cofactors of CmaA1 taken from molecular dynamics (MD) simulations trajectories of CmaA1. The screening abilities of these models were validated by screening 23 inhibitors and 1398 non-inhibitors of CmaA1. A VS protocol was designed with four levels of screening i.e., ligand-based pharmacophore screening, structure-based pharmacophore screening, docking and absorption, distribution, metabolism, excretion and the toxicity (ADMET) filters.
Keep Reading...Structural and Functional Diversities of the Hexadecahydro‐1H‐cyclopenta [a] phenanthrene Framework, a Ubiquitous Scaffold in Steroidal Hormones
C Choudhury, UD Priyakumar, GN Sastry,

Hexadecahydro-1H-cyclopenta[a]phenanthrene framework (HHCPF) has been considered as one of the privileged scaffolds due to its versatile presence in many biologically essential molecules. In our quest to unravel the privileged nature of this framework, we undertook a systematic analysis of target binding and Absorption, Distribution, Metabolism, Elimination, Toxicity (ADMET)/physicochemical properties of 110 drugs containing HHCPF reported in DrugBank. Effect of number and positions of double bonds in the framework and substitutions at each carbon position on the target selectivity as well as drug like properties of these drugs were studied. Fifteen different scaffolds based on the numbers and positions of double bonds in the HHCPF were identified among these drugs. The optimum number of double bonds present in the HHCPF scaffolds was observed to be one to three, and one particular positional isomer is predominant among many scaffolds with same numbers of double bonds.
Keep Reading...Ability of density functional theory methods to accurately model the reaction energy pathways of the oxidation of CO on gold cluster: A benchmark study
S Gurtu, S Rai, M Ehara, UD Priyakumar
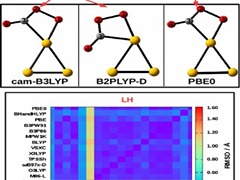
Gold clusters are currently regarded as new-generation catalysts owing to their exceptional efficiency in accelerating several classes of reactions. Density functional theory (DFT) is the method of choice for the investigation of energy pathways of reactions assisted by metal nanoparticles due to their computational efficiency. However, the reliability of such theoretical studies depends to a large extent on the choice of the DFT functional used. In the present work, the performance of a series of DFT-based functionals to accurately model the prototypical CO oxidation reaction catalyzed by a Au3Au3 cluster has been examined by comparing the results with those obtained from high-level ab initio CCSD(T) method.
Keep Reading...Sumoylation of Sir2 differentially regulates transcriptional silencing in yeast
A Hannan, NM Abraham, S Goyal,I Jamir, UD Priyakumar, K Mishra
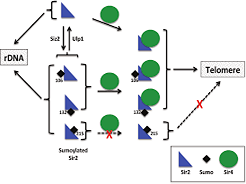
Silent information regulator 2 (Sir2), the founding member of the conserved sirtuin family of NAD(+)-dependent histone deacetylase, regulates several physiological processes including genome stability, gene silencing, metabolism and life span in yeast. Within the nucleus, Sir2 is associated with telomere clusters in the nuclear periphery and rDNA in the nucleolus and regulates gene silencing at these genomic sites. How distribution of Sir2 between telomere and rDNA is regulated is not known.
Keep Reading...Modeling the structure of SARS 3a transmembrane protein using a minimum unfavorable contact approach
S Ramakrishna, S Padhi, UD Priyakumar
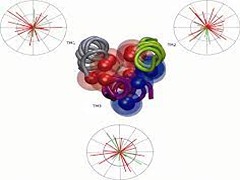
3a is an accessory protein from SARS coronavirus that is known to play a significant role in the proliferation of the virus by forming tetrameric ion channels. Although the monomeric units are known to consist of three transmembrane (TM) domains, there are no solved structures available for the complete monomer. The present study proposes a structural model for the transmembrane region of the monomer by employing our previously tested approach, which predicts potential orientations of TM α-helices by minimizing the unfavorable contact surfaces between the different TM domains. The best model structure comprising all three α-helices has been subjected to MD simulations to examine its quality. The TM bundle was found to form a compact and stable structure with significant intermolecular interactions. The structural features of the proposed model of 3a account for observations from previous experimental investigations on the activity of the protein.
Keep Reading...Molecular dynamics study of the structure, flexibility, and hydrophilicity of PETIM dendrimers: a comparison with PAMAM dendrimers
S Kanchi, G Suresh, UD Priyakumar, KG Ayappa, PK Maiti.
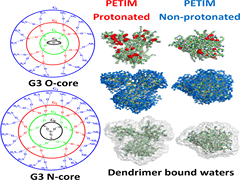
A new class of dendrimers, the poly(propyl ether imine) (PETIM) dendrimer, has been shown to be a novel hyperbranched polymer having potential applications as a drug delivery vehicle. Structure and dynamics of the amine terminated PETIM dendrimer and their changes with respect to the dendrimer generation are poorly understood. Since most drugs are hydrophobic in nature, the extent of hydrophobicity of the dendrimer core is related to its drug encapsulation and retention efficacy. In this study, we carry out fully atomistic molecular dynamics (MD) simulations to characterize the structure of PETIM (G2–G6) dendrimers in salt solution as a function of dendrimer generation at different protonation levels. Structural properties such as radius of gyration (Rg), radial density distribution, aspect ratio, and asphericity are calculated. In order to assess the hydrophilicity of the dendrimer, we compute the number of bound water molecules in the interior of dendrimer as well as the number of dendrimer–water hydrogen bonds. We conclude that PETIM dendrimers have relatively greater hydrophobicity and flexibility when compared with their extensively investigated PAMAM counterparts.
Keep Reading...Atomistic details of the molecular recognition of DNA-RNA hybrid duplex by ribonuclease H enzyme
G Suresh, UD Priyakumar,
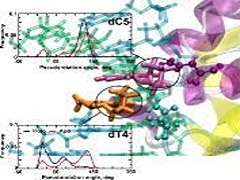
Bacillus halodurans (Bh) ribonuclease H (RNase H) belongs to the nucleotidyl-transferase (NT) superfamily and is a prototypical member of a large family of enzymes that use two-metal ion (Mg2+ or Mn2+) catalysis to cleave nucleic acids. Long timescale molecular dynamics simulations have been performed on the BhRNase H-DNA-RNA hybrid complex and the respective monomers to understand the recognition mechanism, conformational preorganization, active site dynamics and energetics involved in the complex formation. Several structural and energetic analyses were performed and significant structural changes are observed in enzyme and hybrid duplex during complex formation.
Keep Reading...Inclusion of methoxy groups inverts the thermodynamic stabilities of DNA– RNA hybrid duplexes: A molecular dynamics simulation study
G Suresh, UD Priyakumar
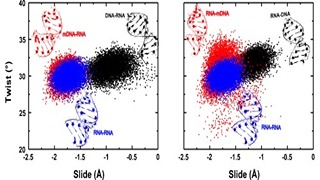
Modified nucleic acids have found profound applications in nucleic acid based technologies such as antisense and antiviral therapies. Previous studies on chemically modified nucleic acids have suggested that modifications incorporated in furanose sugar especially at 2'-position attribute special properties to nucleic acids when compared to other modifications. 2'-O-methyl modification to deoxyribose sugars of DNA-RNA hybrids is one such modification that increases nucleic acid stability and has become an attractive class of compounds for potential antisense applications. It has been reported that modification of DNA strands with 2'-O-methyl group reverses the thermodynamic stability of DNA-RNA hybrid duplexes.
Keep Reading...Ion hydration dynamics in conjunction with a hydrophobic gating mechanism regulates ion permeation in p7 viroporin from hepatitis C virus
S Padhi, UD Priyakumar
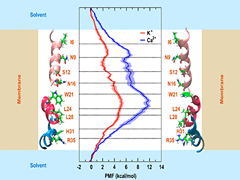
The selectivity of the p7 channel from hepatitis C virus (HCV) toward K+ over Ca2+ has made the channel an intriguing system for investigating ion permeation. The present study employs umbrella sampling free energy calculations to investigate the atomistic details of cation conduction through the channel. The free energy profiles suggest that the energy barrier for Ca2+ conduction is higher than that for K+ conduction by about 4.5 kcal/mol, thus explaining the selectivity exhibited by the channel toward K+.
Keep Reading...Small-molecule inhibitors of ERK-mediated immediate early gene expression and proliferation of melanoma cells expressing mutated BRaf
R Samadani, J Zhang, A Brophy, T Oashi, UD Priyakumar, EP Raman, AD MacKerell Jr., P Shapiro.
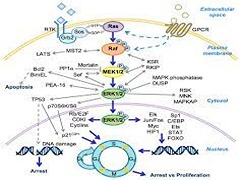
Constitutive activation of the extracellular-signal-regulated kinases 1 and 2 (ERK1/2) are central to regulating the proliferation and survival of many cancer cells. The current inhibitors of ERK1/2 target ATP binding or the catalytic site and are therefore limited in their utility for elucidating the complex biological roles of ERK1/2 through its phosphorylation and regulation of over 100 substrate proteins.
Keep Reading...Dynamics based pharmacophore models for screening potential inhibitors of mycobacterial cyclopropane synthase
C Choudhury, UD Priyakumar, GN Sastry,
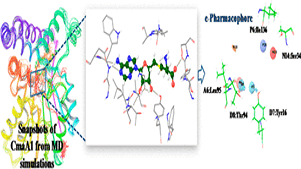
The therapeutic challenges in the treatment of tuberculosis demand multidisciplinary approaches for the identification of potential drug targets as well as fast and accurate techniques to screen huge chemical libraries. Mycobacterial cyclopropane synthase (CmaA1) has been shown to be essential for the survival of the bacteria due to its critical role in the synthesis of mycolic acids.
Keep Reading...Prediction of the structures of helical membrane proteins based on a minimum unfavorable contacts approach
S Padhi, S Ramakrishna, UD Priyakumar
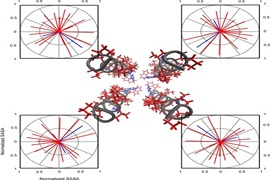
An understanding of structure-function relationships of membrane proteins continues to be a challenging problem, owing to the difficulty in obtaining their structures experimentally. This study suggests a method for modeling membrane protein structures that can be used to generate a reliable initial conformation prior to the use of other approaches for sampling conformations.
Keep Reading...Dispersion interactions between urea and nucleobases contribute to the destabilization of RNA by urea in aqueous solution
K Kasavajhala, S Bikkina, I Patil, AD MacKerell Jr, UD Priyakumar
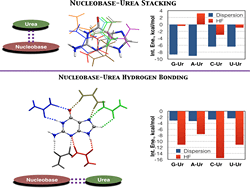
Urea has long been used to investigate protein folding and, more recently, RNA folding. Studies have proposed that urea denatures RNA by participating in stacking interactions and hydrogen bonds with nucleic acid bases. In this study, the ability of urea to form unconventional stacking interactions with RNA bases is investigated using ab initio calculations (RI-MP2 and CCSD(T) methods with the aug-cc-pVDZ basis set).
Keep Reading...Nucleobases tagged to gold nanoclusters cause a mechanistic crossover in the oxidation of CO
S Rai, M Ehara, UD Priyakumar
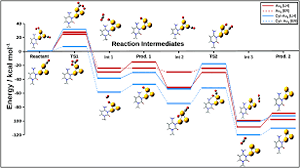
DNA is considered as a programmable building block for the assembly of nanomaterials that play a significant role in modern day nanotechnology and catalysis. In this work, density functional theory (DFT) is used to explore the possible application of complexes of DNA bases (adenine (A), thymine (T), guanine (G), and cytosine (C)) and their size expanded (x) counterparts with Au3 gold clusters as a model catalyst system for oxidation of CO to CO2.
Keep Reading...Binding to gold nanoclusters alters the hydrogen bonding interactions and electronic properties of canonical and size-expanded DNA base pairs
S Rai, H Singh, UD Priyakumar

DNA molecules tagged to metal nanoparticles, especially gold nanoparticles (AuNPs), have been shown to have potential applications in the design and fabrication of novel electronic nano-devices, but the binding mechanism between gold nanoparticles and DNA bases and its implications are not completely understood.
Keep Reading...Double zipper helical assembly of deoxyoligonucleotides: mutual templating and chiral imprinting to form hybrid DNA ensembles
N Narayanaswamy, G Suresh, UD Priyakumar, T Govindaraju
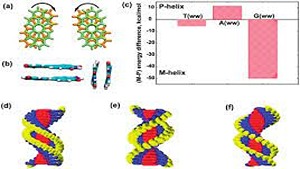
Herein, the conventional and unconventional hydrogen bonding potential of adenine in APA for double zipper helical assembly of deoxyoligonucleotides is demonstrated under ambient conditions.
Keep Reading...Molecular dynamics investigation of the active site dynamics of mycobacterial cyclopropane synthase during various stages of the cyclopropanation process
C Choudhury, UD Priyakumar, GN Sastry
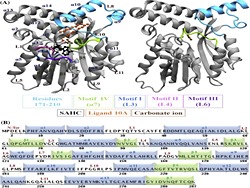
Mycobacterial cyclopropane synthase 1 (CmaA1) is one of the most important drug targets in anti tuberculosis drug discovery as it is responsible for cis-cyclopropanation at the distal position of unsaturated mycolates, which is an essential step for the pathogenicity, persistence and drug resistance.
Keep Reading...Atomistic investigation of the effect of incremental modification of deoxyribose sugars by locked nucleic Acid (β-d-LNA and α-l-LNA) moieties on the structures and thermodynamics of DNA-RNA hybrid duplexes,
G Suresh, UD Priyakumar
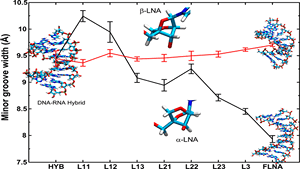
Chemically modified oligonucleotides offer many possibilities in utilizing their special features for a vast number of applications in nucleic acid based therapies and synthetic molecular biology. Locked nucleic acid analogues (α-/β-LNA) are modifications having an extra ring of 2′-O,4′-C-methylene group in the furanose sugar.
Keep Reading...Atomistic detailed mechanism and weak cation-conducting activity of HIV-1 Vpu revealed by free energy calculations
S Padhi, RR Burri, S Jameel, UD Priyakumar
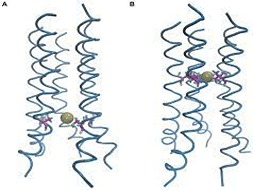
The viral protein U (Vpu) encoded by HIV-1 has been shown to assist in the detachment of virion particles from infected cells. Vpu forms cation-specific ion channels in host cells, and has been proposed as a potential drug target. An understanding of the mechanism of ion transport through Vpu is desirable, but remains limited because of the unavailability of an experimental structure of the channel.
Keep Reading...Modulation of structural, energetic and electronic properties of DNA and size- expanded DNA bases upon binding to gold clusters
S Rai, S Ranjan, H Singh, UD Priyakumar,
Gold cluster–nucleobase complexes have potential applications in designing and fabrication of novel electronic nano-devices, and there has been a surge in research activities in this area recently.
Keep Reading...DNA–RNA hybrid duplexes with decreasing pyrimidine content in the DNA strand provide structural snapshots for the A-to B-form conformational transition of nucleic acids.
G Suresh, UD Priyakumar
DNA–RNA hybrids are heterogeneous nucleic acid duplexes consisting of a DNA strand and a RNA strand, and are formed as key intermediates in many important biological processes. They serve as substrates for the RNase H enzymatic activity, which has been exploited for several biomedical technologies such as antiviral and antisense therapies. To understand the relation of structural properties with the base composition in DNA–RNA hybrids, molecular dynamics (MD) simulations were performed on selected model systems by systematically varying the deoxypyrimidine (dPy) content from 0 to 100% in the DNA strand.
Keep Reading...Solvent‐ Induced Helical Assembly and Reversible Chiroptical Switching of Chiral Cyclic‐Dipeptide‐ Functionalized Naphthalenediimides
S Manchineella, V Prathyusha, UD Priyakumar, T Govindaraju,
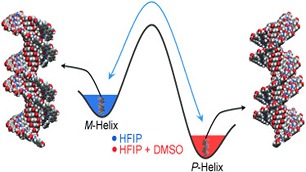
Mastering the handedness: Understanding the roles of various parameters in orchestrating the preferential chiral molecular organization in supramolecular self-assembly processes is of great significance in designing novel molecular functional systems. This study emphasizes the role of cyclic dipeptide chiral auxiliaries on the solvent-induced helical assembly and reversible chiroptical switching of naphthalenediimides (see figure: HFIP=1,1,1,3,3,3-hexafluoroisopropanol).
Keep Reading...Structures, Dynamics, and Stabilities of Fully Modified Locked Nucleic Acid (β-d-LNA and α-l-LNA) Duplexes in Comparison to Pure DNA and RNA Duplexes
G Suresh, UD Priyakumar,
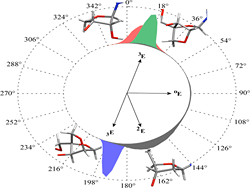
Locked nucleic acid (LNA) is a chemical modification which introduces a −O–CH2– linkage in the furanose sugar of nucleic acids and blocks its conformation in a particular state. Two types of modifications, namely, 2′-O,4′-C-methylene-β-d-ribofuranose (β-d-LNA) and 2′-O,4′-C-methylene-α-l-ribofuranose (α-l-LNA), have been shown to yield RNA and DNA duplex-like structures, respectively. LNA modifications lead to increased melting temperatures of DNA and RNA duplexes, and have been suggested as potential therapeutic agents in antisense therapy. In this study, molecular dynamics (MD) simulations were performed on fully modified LNA duplexes and pure DNA and RNA duplexes sharing a similar sequence to investigate their structure, stabilities, and solvation properties. Both LNA duplexes undergo unwinding of the helical structure compared to the pure DNA and RNA duplexes. Though the α-LNA substituent has been proposed to mimic deoxyribose sugar in its conformational properties, the fully modified duplex was found to exhibit unique structural and dynamic properties with respect to the other three nucleic acid structures.
Keep Reading...Synthesis and Reactivity Studies of Dicationic Dihydrogen Complexes Bearing Sulfur‐Donor Ligands: A Combined Experimental and Computational Study
T Gandhi, S Rajkumar, V Prathyusha, UD Priyakumar
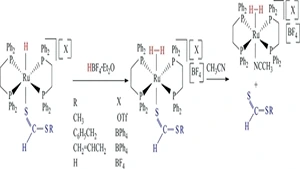
A series of dihydrogen complexes trans-[Ru(η2-H2){SC(SR)H}(dppe)2][X][BF4] (R = CH3, X = OTf; R = C6H5CH2, X = BPh4; R = H2C=CHCH2, X = BPh4; dppe = Ph2PCH2CH2PPh2) bearing sulfur-donor ligands has been synthesized by protonation of the (alkyl dithioformate)hydrido complexes trans-[Ru(H){SC(SR)H}(dppe)2][X] by using HBF4·Et2O.
Keep Reading...Role of conformational properties on the transannular Diels–Alder reactivity of macrocyclic trienes with varying linker lengths
V Prathyusha, UD Priyakumar,
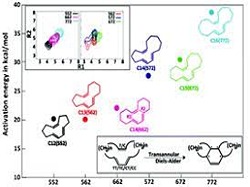
The effect of the linker length (–CH2–) connecting the diene and the dienophile in macrocyclic trienes on their transannular Diels–Alder (TADA) reactivity has been investigated using density functional theory (DFT) calculations. The relationship between the conformational properties of these reactants and their reaction energy barriers was examined and a quantitative relationship has been obtained. The transition state energy barriers were found to increase with an increase in the linker length, which is in contrast to the expected trend.
Keep Reading...Molecular dynamics simulations reveal the HIV-1 Vpu transmembrane protein to form stable pentamers
S Padhi, N Khan, S Jameel, UD Priyakumar,
The human immunodeficiency virus type I (HIV-1) Vpu protein is 81 residues long and has two cytoplasmic and one transmembrane (TM) helical domains. The TM domain oligomerizes to form a monovalent cation selective ion channel and facilitates viral release from host cells. Exactly how many TM domains oligomerize to form the pore is still not understood, with experimental studies indicating the existence of a variety of oligomerization states. In this study, molecular dynamics (MD) simulations were performed to investigate the propensity of the Vpu TM domain to exist in tetrameric, pentameric, and hexameric forms. Starting with an idealized α-helical representation of the TM domain, a thorough search for the possible orientations of the monomer units within each oligomeric form was carried out using replica-exchange MD simulations in an implicit membrane environment. Extensive simulations in a fully hydrated lipid bilayer environment on representative structures obtained from the above approach showed the pentamer to be the most stable oligomeric state, with interhelical van der Waals interactions being critical for stability of the pentamer. Atomic details of the factors responsible for stable pentamer structures are presented. The structural features of the pentamer models are consistent with existing experimental information on the ion channel activity, existence of a kink...
Keep Reading...Crenarchaeal chromatin proteins Cren7 and Sul7 compact DNA by inducing rigid bends
RPC Driessen, H Meng, G Suresh, R Shahapure, G Lanzani, UD Priyakumar
Archaeal chromatin proteins share molecular and functional similarities with both bacterial and eukaryotic chromatin proteins. These proteins play an important role in functionally organizing the genomic DNA into a compact nucleoid. Cren7 and Sul7 are two crenarchaeal nucleoid-associated proteins, which are structurally homologous, but not conserved at the sequence level. Co-crystal structures have shown that these two proteins induce a sharp bend on binding to DNA. In this study, we have investigated the architectural properties of these proteins using atomic force microscopy, molecular dynamics simulations and magnetic tweezers. We demonstrate that Cren7 and Sul7 both compact DNA molecules to a similar extent. Using a theoretical model, we quantify the number of individual proteins bound to the DNA as a function of protein concentration and show that forces up to 3.5 pN do not affect this binding. Moreover, we investigate the flexibility of the bending angle induced by Cren7 and Sul7 and show that the protein–DNA complexes differ in flexibility from analogous bacterial and eukaryotic DNA-bending proteins.
Keep Reading...Inter-versus intra-molecular cyclization of tripeptides containing tetrahydrofuran amino acids: a density functional theory study on kinetic control
NVS Kumar, UD Priyakumar, H Singh, S Roy, TK Chakraborty
Density functional B3LYP method was used to investigate the preference of intra- and inter-molecular cyclizations of linear tripeptides containing tetrahydrofuran amino acids. Two distinct model pathways were conceived for the cyclization reaction, and all possible transition states and intermediates were located. Analysis of the energetics indicate intermolecular cyclization being favored by both thermodynamic and kinetic control. Geometric and NBO analyses were performed to explain the trends obtained along both the reaction pathways. Conceptual density functional theory-based reactive indices also show that reaction pathways leading to intermolecular cyclization of the tripeptides are relatively more facile compared to intramolecular cyclization.
Keep Reading...Transannular Diels–Alder reactivities of 14-membered macrocylic trienes and their relationship with the conformational preferences of the reactants: A combined quantum chemical and molecular dynamics study
V Prathyusha, S Ramakrishna, UD Priyakumar
Transannular Diels–Alder (TADA) reactions that occur between the diene and dienophile moieties located on a single macrocyclic triene molecule have been recognized as effective synthetic routes toward realizing complex tricyclic molecules in a single step. In this paper, we report a comprehensive study on the TADA reactions of 14-membered cyclic triene macrocycles to yield A.B.C[6.6.6] tricycles using quantum chemical methods and using classical molecular dynamics simulations. A benchmark study has been performed to examine the reliability of the commonly used ab initio methods and hybrid density functional levels of theory in comparison with results from CCSD(T) calculations to accurately model TADA reactions. The energy barriers obtained using the M06-2X functional were found to be in quantitative agreement with the CCSD(T) level of theory using a reasonably large basis set. Conformational properties of the reactants have been systematically studied using extensive molecular dynamics (MD) simulations. For this purpose, model systems were conceived, and force field parameters corresponding to the dihedral terms in the potential energy function were obtained. Linear relationship between the activation energies corresponding to the TADA reactions and the probability of finding the reactant in certain conformational states was obtained. A clustering method along with optimizations at the molecular mechanics and density functional M06-2X...
Keep Reading...Computational investigation of the effect of thermal perturbation on the mechanical unfolding of titin I27
Bung, UD Priyakumar
The emergence of single-molecule force measurement experiments has facilitated a better understanding of protein folding pathways and the thermodynamics involved. Computational methods such as steered molecular dynamics (SMD) simulations are helpful in providing atomistic level information on the unfolding pathways. Recent experimental studies have showed that combinations of single-molecule experiments with traditional methods such as chemical and/or thermal denaturation yield additional insights into the folding phenomenon. In this study, we report results from extensive computations (a total of about 60 SMD simulations with a total length of about 0.4 μs) that address the effect of thermal perturbation on the mechanical stability of the I27 domain of the protein titin. A wide range of temperatures (280-340 K) were considered for the pulling, which was done at both constant velocity and constant force using SMD simulations. Good agreement with experimental data, such as for the trends in changes in average force and the maximum force with respect to the temperature, was obtained. This study identifies two competing pathways for the mechanical unfolding of I27, and illustrates the significance of combining various techniques to examine protein folding.
Keep Reading...Role of Hydrophobic Core on the Thermal Stability of Proteins—Molecular Dynamics Simulations on a Single Point Mutant of Sso7d
UD Priyakumar
The role of salt bridges in chromatin protein Sso7d, from S. solfataricus has previously been shown to be crucial for its unusual high thermal stability. Experimental studies have shown that single site mutation of Sso7d (F31A) leads to a substantial decrease in the thermal stability of this protein due to distortion of the hydrophobic core. In the present study, we have performed a total of 0.2 s long molecular dynamics (MD) simulations on F31A at room temperature, and at 360 K, close to the melting temperature of the wild type (WT) protein to investigate the role of hydrophobic core on protein stability. Sso7d-WT was shown to be stable at both 300 and 360 K; however, F31A undergoes denaturation at 360 K, consistent with experimental results. The structural and energetic properties obtained using the analysis of MD trajectories indicate that the single mutation results in high flexibility of the protein, and loosening of intramolecular interactions. Correlation between the dynamics of the salt bridges with the structural transitions and the unfolding pathway indicate the importance of both salt bridges and hydrophobic in effecting thermal stability of proteins in general.
Keep Reading...Characterization of ERK docking domain inhibitors that induce apoptosis by targeting Rsk-1 and caspase-9
SR Boston, R Deshmukh, S Strome, UD Priyakumar, AD MacKerell, P Shapiro,

The extracellular signal-regulated kinase-1 and 2 (ERK1/2) proteins play an important role in cancer cell proliferation and survival. ERK1/2 proteins also are important for normal cell functions. Thus, anti-cancer therapies that block all ERK1/2 signaling may result in undesirable toxicity to normal cells. As an alternative, we have used computational and biological approaches to identify low-molecular weight compounds that have the potential to interact with unique ERK1/2 docking sites and selectively inhibit interactions with substrates involved in promoting cell proliferation.
Keep Reading...Impact of 2′‐hydroxyl sampling on the conformational properties of RNA: Update of the CHARMM all‐atom additive force field for RNA, EJ Denning
UD Priyakumar, L Nilsson, AD Mackerell Jr
Here, we present an update of the CHARMM27 all-atom additive force field for nucleic acids that improves the treatment of RNA molecules. The original CHARMM27 force field parameters exhibit enhanced Watson-Crick base pair opening which is not consistent with experiment, whereas analysis of molecular dynamics (MD) simulations show the 2'-hydroxyl moiety to almost exclusively sample the O3' orientation. Quantum mechanical (QM) studies of RNA related model compounds indicate the energy minimum associated with the O3' orientation to be too favorable, consistent with the MD results. Optimization of the dihedral parameters dictating the energy of the 2'-hydroxyl proton targeting the QM data yielded several parameter sets, which sample both the base and O3' orientations of the 2'-hydroxyl to varying degrees. Selection of the final dihedral parameters was based on reproduction of hydration behavior as related to a survey of crystallographic data and better agreement with experimental NMR J-coupling values. Application of the model, designated CHARMM36, to a collection of canonical and noncanonical RNA molecules reveals overall improved agreement with a range of experimental observables as compared to CHARMM27. The results also indicate the sensitivity of the conformational heterogeneity of RNA to the orientation of the 2'-hydroxyl moiety and support a model whereby the 2'-hydroxyl can enhance...
Keep Reading...Molecular simulations on the thermal stabilization of DNA by hyperthermophilic chromatin protein Sac7d, and associated conformational transitions
UD Priyakumar, G Harika, G Suresh
Sac7d belongs to a family of chromosomal proteins, which are crucial for thermal stabilization of DNA at higher growth temperatures. It is capable of binding DNA nonspecifically, and is responsible for the increase in the melting temperature of DNA in the bound form up to 85 °C. Molecular dynamics (MD) simulations were performed at different temperatures on two protein−DNA complexes of Sac7d. Various structural and energetic parameters were calculated to examine the DNA stability and to investigate the conformational changes in DNA and the protein−DNA interactions. Room temperature simulations indicated very good agreement with the experimental structures. The protein structure is nearly unchanged at both 300 and 360 K, and only up to five base pairs of the DNA are stabilized by Sac7d at 360 K. However, the MD simulations on DNA alone systems show that they lose their helical structures at 360 K further supporting the role of Sac7d in stabilizing the oligomers. At higher temperatures (420 and 480 K), DNA undergoes denaturation in the presence and the absence of the protein. The DNA molecules were found to undergo B- to A-form transitions consistent with experimental studies, and the extent of these transitions are examined in detail. The extent of sampling B- and A-form...
Keep Reading...Atomistic Details of the Ligand Discrimination Mechanism of SMK/SAM-III Riboswitch
UD Priyakumar
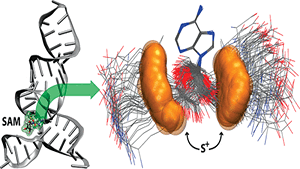
SAM-III riboswitch, involved in regulating sulfur metabolic pathways in lactic acid bacteria, is capable of differentiating S-adenosyl-l-methionine (SAM) from its structurally similar analogue S-adenosyl-l-homocysteine (SAH). Atomic level understanding of the ligand recognition mechanism of riboswitches is essential for understanding their structure−function relationships in general. In the present study, we have employed molecular dynamics (MD) simulations on five model systems to elucidate the discrimination mechanism adopted by the SAM-III riboswitch that enables differential binding of SAM with respect to SAH. The structures of the binding pocket of the riboswitch and the modes of binding of the adenine moiety of SAM obtained from the MD simulations are similar to the experimental structure. However, MD simulations of the riboswitch−SAH complexes lead to partial unbinding of the ligand and structural changes in the RNA binding pocket. Detailed analyses were performed to examine the structural and energetic factors involved in such a differentiation. The calculations reveal a novel mechanism by which the aptamer domain specifically recognizes the adenine moiety of SAM/SAH, but SAM is better stabilized in the binding pocket due to nonspecific electrostatic interactions involving the sulfonium group. Additionally, the results support less dependence of the ligand conformation in the bound form on the effective binding of SAM to the riboswitch.
Keep Reading...Structural and energetic determinants of thermal stability and hierarchical unfolding pathways of hyperthermophilic proteins, Sac7d and Sso7d
UD Priyakumar, S Ramakrishna, KR Nagarjuna, SK Reddy
Identification of the structural and energetic determinants responsible for enhancing the stability of proteins is crucial. Hyperthermophilic proteins are naturally occurring proteins that exhibit high thermal stability and are good candidates for the investigation and understanding of structure−stability relationships. Sac7d from Sulfolobus acidocaldarius and Sso7d from Sulfolobus solfactaricus are two homologous hyperthermophilic proteins that were shown to be quite stable at high temperatures. Molecular dynamics simulations at the nanosecond time scale at different temperatures were performed to examine the factors affecting their stability. The three-dimensional structures of these proteins were observed to be similar to the experimental structure at 300 and 360 K but were found to undergo denaturation at 500 K. Both proteins exhibit similar unfolding pathways that correlates well with the calculated intermolecular interaction energies. The differential dynamic behaviors of these molecules at different temperatures were examined. Structural and energetic analysis of the contributions of salt bridges indicates a stabilizing effect at higher temperatures. However, the lifetimes of the salt bridges were found to be quite short, and several new salt bridges formed at 500 K supporting previous studies that the desolvation penalty due to the formation of salt bridges decreases at elevated temperatures. Hydrophobic interactions, which decrease with increase in temperature, were also found to be...
Keep Reading...Role of the adenine ligand on the stabilization of the secondary and tertiary interactions in the adenine riboswitch
UD Priyakumar, AD MacKerell Jr,
Riboswitches are RNA-based genetic control elements that function via a conformational transition mechanism when a specific target molecule binds to its binding pocket. To facilitate an atomic detail interpretation of experimental investigations on the role of the adenine ligand on the conformational properties and kinetics of folding of the add adenine riboswitch, molecular dynamics (MD) simulations were performed in both the presence and absence of the ligand. In the absence of ligand, structural deviations were observed in the J23 junction and the P1 stem. Destabilization of the P1 stem in the absence of ligand involves the loss of direct stabilizing interactions with the ligand, with additional contributions from the J23 junction region. The J23 junction of the riboswitch is found to be more flexible and the tertiary contacts among the junction regions are altered in the absence of the adenine ligand; results suggest that the adenine ligand associates and dissociates from the riboswitch in the vicinity of J23. Good agreement was obtained with the experimental data with the results indicating dynamic behavior of the adenine ligand on the nanosecond timescale to be associated with the dynamic behavior of hydrogen bonding with the riboswitch. Results also predict that direct interactions of the adenine ligand with U74 of the...
Keep Reading...Urea destabilizes RNA by forming stacking interactions and multiple hydrogen bonds with nucleic acid bases
UD Priyakumar, C Hyeon, D Thirumalai, AD MacKerell Jr
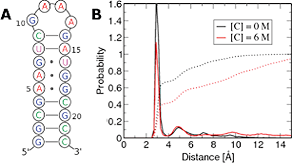
Urea titration of RNA by urea is an effective approach to investigate the forces stabilizing this biologically important molecule. We used all atom molecular dynamics simulations using two urea force fields and two RNA constructs to elucidate in atomic detail the destabilization mechanism of folded RNA in aqueous urea solutions. Urea denatures RNA by forming multiple hydrogen bonds with the RNA bases and has little influence on the phosphodiester backbone. Most significantly we discovered that urea engages in stacking interactions with the bases. We also estimate, for the first time, the m-value for RNA, which is a measure of the strength of urea-RNA interactions. Our work provides a conceptual understanding of the mechanism by which urea enhances RNA folding rates.
Keep Reading...Molecular modeling of base flipping
UD Priyakumar, AD MacKerell Jr
Carrying out chemistry on the bases of DNA, necessary for biological processes such as methylation or repair, requires flipping the base into an accessible position. In this work, molecular dynamics simulations are used to generate a free energy profile for flipping a cytosine base out of its helical stack in double-stranded DNA. The results shed light on the mechanics of this process by comparing routes for base flipping via the minor and major grooves.
Keep Reading...Atomic Detail Investigation of the Structure and Dynamics of DNA• RNA Hybrids: A Molecular Dynamics Study
UD Priyakumar, AD MacKerell Jr
DNA•RNA hybrid duplexes are biologically important molecules and are shown to have potential therapeutic properties. To investigate the relationship between structures, energetics, solvation and RNase H activity of hybrid duplexes in comparison with pure DNA and RNA duplexes, a molecular dynamics study using the CHARMM27 force field was undertaken. The structural properties of all four nucleic acids considered are in very good agreement with the experimental data. The backbone dihedral angles and the puckering of the (deoxy)ribose indicate that the purine rich strands retain their A-/B-like properties but the pyrimidine rich DNA strand undergoes A−B conformational transitions. The minor groove widths of the hybrid structures are narrower than those in the RNA duplex, a requirement for RNase H binding. In addition, sampling of noncanonical phosphodiester backbone dihedrals by the DNA strands, differential solvation properties and helical properties, most notably rise, are suggested to contribute to hybrids being RNase H substrates. Differential RNase H activity toward hybrids containing purine versus pyrimidine rich RNA strands is suggested to be due to sampling of values of the phosphodiester backbone dihedrals in the DNA strands. Notably, the present results indicate that hybrids have decreased flexibility as compared to RNA, in contrast to previous reports.
Keep Reading...Computational approaches for investigating base flipping in oligonucleotides
UD Priyakumar, AD MacKerell,
NMR imino proton exchange experiments on duplex DNA primarily monitor the opening of purine bases
UD Priyakumar, AD MacKerell,
Molecular dynamics simulations were performed to investigate GC and AT base opening events in DNA. Calculated equilibrium constants between the base open (or flipped) and closed states were shown to be in good agreement with experimental data from NMR imino proton exchange experiments. Analysis of the computed results indicates that the equilbrium constants are dominated by the opening of the A and G bases in the AT and GC base pairs, respectively. Thus, the present results predict that NMR imino proton exchange experiments of base opening are primarily monitoring the opening of purine bases.
Keep Reading...Base flipping in a GCGC containing DNA dodecamer: A comparative study of the performance of the nucleic acid force fields, CHARMM, AMBER, and BMS
UD Priyakumar, AD MacKerell,
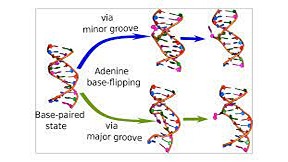
The improving quality of empirical force field parameters along with other methodological improvements and ever increasing computational resources have lead to more reliable computations on biological macromolecules. In the case of oligonucleotides, three force fields, namely CHARMM27, AMBER4.1, and BMS, have been developed and are widely used by the simulation community. Testing of these force fields to date has primarily focused on their treatment of the canonical forms of DNA and RNA. However, many biological functions of oligonucleotides involve significant variation of their structures from the canonical forms. In the present work, the three force fields are evaluated via computation of potentials of mean force (PMF) of the base flipping process in a DNA dodecamer, 5'-GTCAGCGCATGG-3'. Results are compared with available experimental data on the equilibrium between the opened and closed (i.e. Watson-Crick base paired) state of the underlined C and its WC partner G. Quantitative analysis shows CHARMM to be in the best agreement with experiment, closely followed by AMBER with BMS in the poorest agreement. Various components contributing to the change in the free energy such as base pair interactions, stacking interactions, solvation effects, and intrinsic potential energy changes were evaluated and compared. The results indicate that while all three force fields reasonably...
Keep Reading...A lipophilic hexaporphyrin assembly supported on a stannoxane core
V Chandrasekhar, S Nagendran, R Azhakar, MR Kumar, A Srinivasan, UD Priyakumar, GN Sastry
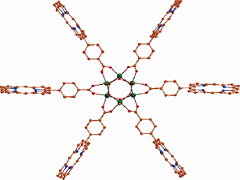
Lipophilic hexaporphyrin free-base and copper-metalated assemblies supported on a Sn6O6 core have been synthesized and characterized. The nuclease activity of the copper derivative has been studied.
Keep Reading...Conformational determinants of tandem GU mismatches in RNA: insights from molecular dynamics simulations and quantum mechanical calculations
Y Pan, UD Priyakumar, AD MacKerell Jr
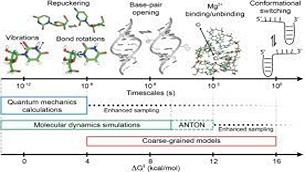
Structure and energetic properties of base pair mismatches in duplex RNA have been the focus of numerous investigations due to their role in many important biological functions. Such efforts have contributed to the development of models for secondary structure prediction of RNA, including the nearest-neighbor model. In RNA duplexes containing GU mismatches, 5'-GU-3' tandem mismatches have a different thermodynamic stability than 5'-UG-3' mismatches. In addition, 5'-GU-3' mismatches in some sequence contexts do not follow the nearest-neighbor model for stability. To characterize the underlying atomic forces that determine the structural and thermodynamic properties of GU tandem mismatches, molecular dynamics (MD) simulations were performed on a series of 5'-GU-3' and 5'-UG-3' duplexes in different sequence contexts. Overall, the MD-derived structural models agree well with experimental data, including local deviations in base step helicoidal parameters in the region of the GU mismatches and the model where duplex stability is associated with the pattern of GU hydrogen bonding. Further analysis of the simulations, validated by data from quantum mechanical calculations, suggests that the experimentally observed differences in thermodynamic stability are dominated by GG interstrand followed by GU intrastrand base stacking interactions that dictate the one versus two hydrogen bonding scenarios for the GU pairs. In addition, the inability of...
Keep Reading...Exploration of C6H6 potential energy surface: A computational effort to unravel the relative stabilities and synthetic feasibility of new benzene isomers
TC Dinadayalane, UD Priyakumar, GN Sastry
Ab initio (MP2, CCSD(T)) and hybrid density functional theory (B3LYP) calculations with up to triple-ζ basis set were done to locate all possible minima, where each carbon in the molecule is tetracoordinate, on the C6H6 potential energy surface. The search was initiated with a total of 218 structures, and in few cases, geometrical and stereoisomers were considered. The exhaustive study on all these topological structures resulted in a total of 263 stationary points on the C6H6 potential energy surface. The B3LYP level characterizes 209 as minima, 31 as transition states, 8 as second-order, 7 as third-order, and 1 as fourth-order saddle points. The remaining 7 structures could be located as stationary points only at the MP2 level. The molecules were classified into acyclic, monocyclic, bicyclic, tricyclic, and tetracyclic. The acyclic isomers fall within a range of 60−80 kcal/mol higher in energy compared to benzene. Among the cyclic structures, the range of relative stabilities of minima is larger, viz., monocyclic (31−146 kcal/mol), bicyclic (72−159 kcal/mol), tricyclic (72−300 kcal/mol), and tetracyclic (102−156 kcal/mol). Strain due to small three and four membered rings and constrained double and triple bonds control the relative stabilities of these isomers. The computed isomers exhibit various novel bonding modes for carbon, namely, planar tetracoordinate, hypervalent,...
Keep Reading...C21H9Z (Z=− 3 to+ 3): a theoretical study on the redox behaviour of C3 symmetric fragment of C60
UD Priyakumar, GN Sastry, G Mehta,
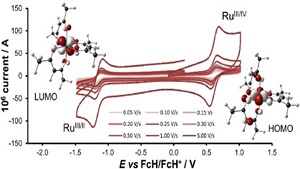
B3LYP/6-31G* calculations were done on seven C21H9Z fragments, where the charge, Z, is varied from −3 to +3; doublet states of the open shell systems and both singlet and triplet states of closed shell species are considered. Among them, only the D3h and C3v forms of the singlet states of C21H93− and C21H91+, and triplet-C21H91− correspond to the bowl-to-bowl inversion transition states and minimum energy structures, respectively. 6-31+G* basis set, which employs a set of diffuse functions on the non-hydrogen atoms, was employed on anionic systems. The rest encountered symmetry breaking problem, which necessitated us to go for lower symmetry structures to obtain the local minima and the bowl-to-bowl inversion transition states. The curvature, ascertained by π-orbital axis vector angles (POAV), is lowest for the trianion and highest for the cations. The triplet states of the closed shell species are found to be competitive in energy with the singlet states when Z=−1 and +3, whereas for Z=−3 and +1, the triplet states lie above the corresponding singlet states over 30 kcal/mol. The inversion barriers vary from 12.2 to 40.2 kcal/mol with C21H93− and C21H92+ having the lowest and highest inversion barriers, respectively. The electron affinities and ionization energies were calculated to assess the electron receiving and donating ability of the C3-fragment in comparison to corannulene. The curvature and redox behavior...
Keep Reading...Facile valence isomerization among bis (silacyclopropenyl), disila (Dewar benzene) and disilabenzvalene
UD Priyakumar, M Punnagai, GN Sastry,
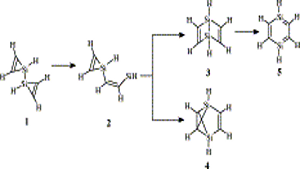
Hybrid density functional theory (B3LYP) and ab initio (CCSD(T)) calculations were performed to explore the reaction energy pathways of the valence isomerizations among the disilabenzene isomers, namely, bis(silacyclopropenyl) (1), disila(Dewar benzene) (3) and disilabenzvalene (4). The computational study predicts that the interconversion of 1 to 3 and 4 occurs spontaneously. However, the interconversion between Dewar benzene (3) and benzvalene (4) analogs is a multistep process involving a new intermediate structure 2a, with energy barriers lying above 30 kcal/mol in energy compared to 3 and 4. Isomerization from bis(silacyclopropenyl) is very facile either to disila(Dewar benzene) or disilabenzvalene.
Keep Reading...A computational study of cation–π interactions in polycyclic systems: exploring the dependence on the curvature and electronic factors
UD Priyakumar, M Punnagai, GPK Mohan, GN Sastry,
Density functional theory (B3LYP) calculations with double and triple-ζ quality basis sets were performed on the Li+ and Na+ π-complexes of corannulene 2, sumanene 3CH2, heterosumanenes 3X, triphenylene 4 and heterotrindenes 5X. The metal ions bind to both convex and concave faces of buckybowls, with a consistent preference to bind to the convex surface by about 1–4 kcal/mol. The metal ion complexation with the π-framework of the central six-membered ring span wider range compared to benzene, indicating the control of size, curvature and electronic perturbations over the strength of cation–π interactions. Computations show that the bowl-to-bowl inversion barriers are only slightly altered upon metal complexation, indicating the continuity of bowl-to-bowl inversion despite metal complexation. We have calculated the binding energies of model systems, triphenylene (4) and heterotrindenes (5X), which indicate that the interaction energies are controlled by electronic factors. While the inversion barrier is dependent mainly on the size of the heteroatom, the extent of binding is independent of the size of the atom or the bowl depth.
Keep Reading...The design of molecules containing planar tetracoordinate carbon,
UD Priyakumar, AS Reddy, GN Sastry,
A system with three contiguous planar tetracoordinate carbons is viable: a computational study on a C6H62= isomer
UD Priyakumar, GN Sastry
Ab initio and density functional theory calculations indicate that a benzene dication isomer (1: C6H62=) with three contiguous planar tetracoordinate carbons is at a minimum on the potential energy surface. The remarkable preference for the planar structure for 1 is traced to the aromatic stabilization present in the three membered ring formed by the three planar tetracoordinate carbon atoms. A novel structure, C6H62=, with three contiguous planar tetracoordinate carbon atoms is proposed as a viable candidate.
Keep Reading...Basis set and method dependence of the relative energies of C2S2H2 isomers
D Vijay, UD Priyakumar, GN Sastry,
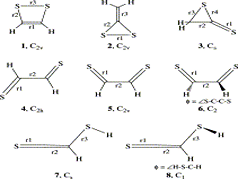
Hartree–Fock, post-Hartree–Fock (MP2 and CCSD(T)) and density functional theory (BLYP, B3LYP, BPW91 and B3PW91) methods were employed to obtain the relative stabilities of eight energetically competitive C2S2H2 isomers. Peculiarly high dependency on the basis set has been observed and the convergence of the relative stability orderings is slow even at the quintuple-ζ levels. Basis sets starting from 6-31G*, 6-311G*, 6-311+G*, 6-311+G**, 6-311++G**, 6-31G(2df), 6-31G(3df), 6-31G(3df,3pd), cc-pVDZ, cc-pVTZ, cc-pVQZ and cc-pV5Z were used in conjunction with various methodologies. A critical comparison of the performance of the density functional methods with MP2, CCSD(T) and G2 methods was made.
Keep Reading...Design of neutral hydrocarbons having a planar tetracoordinate carbon
UD Priyakumar, GN Sastry,
Cyclopropyne (C3H2) and related spiro bicyclic structures show unusual structural preference for the planar tetracoordinate structure over the conventional tetrahedral arrangement. C5H4 has been proposed as the smallest neutral hydrocarbon with planar tetracoordinate carbon . NICS values indicate substantial stabilization of the planar arrangements over the tetrahedral counterparts.
Keep Reading...On the use of NICS criterion to evaluate aromaticity in heteroaromatics involving III and IV row main group elements
A Saieswari, UD Priyakumar, GN Sastry,
Ab initio (HF and MP2) and hybrid density functional theory (B3LYP) calculations were performed on a series of skeletally mono- and di-substituted benzenes with substituents from the II to IV row of the periodic table. The aromaticity in this class of compounds is evaluated based on the nucleus independent chemical shift (NICS) values. Routine classification of 6π-electronic benzenoid systems as aromatic molecules solely based on NICS criterion is questioned in heteroatomics with III and IV row substituents based on the calculated NICS values. However, the NICS criterion is found to be satisfactory in those compounds involving substituents from II row.
Keep Reading...Cation-π interactions of curved polycyclic systems: M+ (M= Li and Na) ion complexation with buckybowls
UD Priyakumar, GN Sastry,
B3LYP/6-311+G** calculations on alkali metal ion (Li+ and Na+) complexation with corannulene and sumanene indicate stronger binding compared to [5]-radialene or benzene. The dependence of binding to the convex and concave site is marginal, albeit the preference was consistent for convex binding in the range of 1–4 kcal/mol. The bowl-to-bowl inversion barriers are only marginally affected, below 2 kcal/mol, by metal ion complexation. Metal ions bind strongly to both π-faces of buckybowls in comparison with benzene and the binding from the convex side is marginally preferred over the concave side.
Keep Reading...The tricyclo [2.1. 0.02, 5] pentan-3-one system: a new probe for the study of π-facial selectivity in nucleophilic additions
G Mehta, SR Singh, UD Priyakumar, GN Sastry,
Monosubstituted tricyclo[2.1.0.02,5]pentan-3-one derivatives have been synthesised and subjected to hydride reduction. Quite unexpectedly and in contrast to earlier observations in related systems, anti-selectivity has been encountered in these substrates. Computational studies employing various models reproduce the observed facial preference and indicate that the causative factor for the anti-preference could be the polarization of the C1C5 strained σ bond
Keep Reading...Silaaromaticity in polycyclic systems: A computational study
DM Dhevi, UD Priyakumar, GN Sastry,
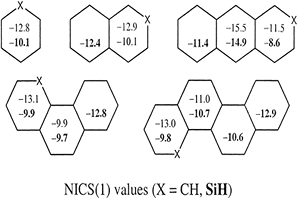
Density functional theory (B3LYP) calculations were performed to examine the effect of Si substitution on the aromaticity of some polycyclic hydrocarbons using geometric criterion (HOMA), isodesmic isomerization reactions, homodesmotic equations, NICS values, chemical hardness, and out-of-plane distortive tendencies. The HOMA values are lower and the NICS values are higher in the Si-substituted rings compared to those in the hydrocarbon counterpart, whereas the homodesmotic equations predict little loss of aromaticity upon Si replacement in polycylic systems. The chemical hardness values decrease and the out-of-plane distortive tendency increases upon silicon substitution. The relative energies of the positional isomers and the causative factors are analyzed. The high reactivity of some silaaromatics toward dimerization is explained based on local softness indices.
Keep Reading...Measures to evaluate heteroaromaticity and their limitations: Story of skeletally substituted benzenes
UD Priyakumar, GN Sastry,
Ab initio HF, MP2, CCSD(T) and hybrid density functional B3LYP calculations were performed on a series of skeletally mono- and di-substituted benzenes, (CH)5Z and (CH)4Z2, Z = C-, N, O+, Si-, P, S+, Ge-, As, Se+, BH-, NH+, AlH-, SiH, PH+, GaH-, GeH and AsH+. Various measures of aromaticity such as the bond length equalization, homodesmic equations, singlet-triplet energy difference (AE s-t), chemical hardness (η) and out-of-plane distortive tendency are critically analysed. The relative energy ordering in skeletally disubstituted benzenes displays trends that are inexplicable based on conventional wisdom. In general, the orthoisomer is found to be the least stable when the substituent is from the second row, whereas if the substituent is from the fourth row, the ortho-isomer is the most stable. Various qualitative arguments, including (a) lone pair-lone pair repulsion, (b) the sum of bond strengths in the twin Kekule forms, and (c) the rule of topological charge stabilization (TCS), are used to explain the observed relative energy trends. The rule of TCS in conjunction with the sum of bond strengths is found to predict the relative energy ordering reasonably well. The reactivity of this class of compounds is assessed based on their singlet-triplet energy differences, chemical hardness and the frequencies corresponding to out-of-plane skeletal...
Keep Reading...The effect of bulky group substitution on the skeleton, geometries, relative energies and the reactivities of silabenzene valence isomers
DM Dhevi, UD Priyakumar, GN Sastry,
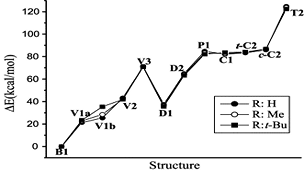
Density functional theory (B3LYP) calculations were performed on the methyl and t-butyl substituted valence isomeric forms of silabenzene. The recently identified 12 valence isomeric minima on the potential energy surface of silabenzene were considered. The calculations reveal that substitution does not affect the skeleton of all the compounds including the unusual structures, V1a and V1b, located on the silabenzene potential energy surface. Similarly, the geometric parameters and relative energies show negligible differences upon substitutions by Me and t-Bu groups except for V1b. Chemical hardness and frontier orbital energy levels are used to assess the reactivity change upon substitution. The present study concludes that the substitution by Me or t-Bu will have only the steric effect and therefore, theoretical models employing the unsubstituted analogues should adequately describe the structural and energetic characteristics of the bulky group substituted silaaromatic compounds.
Keep Reading...Isomers of Disilabenzene (C4Si2H6): A Computational Study
UD Priyakumar, D Saravanan, GN Sastry,
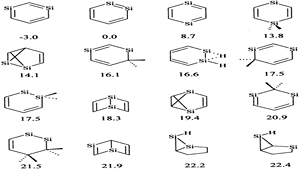
Computational studies using density functional theory (B3LYP) and coupled cluster (CCSD(T)) method were performed on a large number of isomers of disilabenzene, C4Si2H4. In all, 78 stationary points were identified, wherein 61 of them correspond to the minima on the C4Si2H6 potential energy surface. Although the planar forms correspond to the most stable isomers, several unconventional structures are highly competitive in energy. While the framework is an important factor, the substitution pattern seems to be equally important in deciding the relative stabilities of the isomers of disilabenzenes. The relative energies of valence isomeric forms are compared with those of benzene, silabenzene, and diphosphabenzene isomers. The relative energy orderings of all the isomers are analyzed, and conclusions about the stability are drawn. Several H-bridged isomers were computed to be very stable. Several monocyclic six-membered ring structures, where the 6π-aromaticity is disturbed, also indicated the flatness of the potential energy surface of disilabenzene and their propensity toward high reactivity and facile rearrangements.
Keep Reading...π-Facial selectivities in nucleophilic additions to 4-hetero-tricyclo [5.2. 1.02, 6] decan-10-ones and 4-hetero-tricyclo [5.2. 1.02, 6] dec-8-en-10-ones: an experimental and computational study
G Mehta, V Gagliardini, UD Priyakumar, GN Sastry,
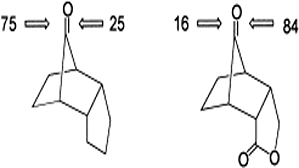
Several endo-tricyclo[5.2.1.02,6]decan-10-ones and endo-tricyclo[5.2.1.02,6]dec-8-en-10-ones with hetero atom modifications at the distal C-4 position have been subjected to hydride reduction. π-Face selectivities in these systems are largely governed by the same electronic factors that were earlier identified in the case of the norbornyl system. A computational study demonstrates good predictability at the semi-empirical level.
Keep Reading...Theoretical study of silabenzene and its valence isomers
UD Priyakumar, GN Sastry,
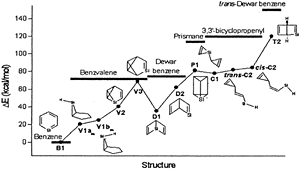
Quantum mechanical calculations at the ab initio (MP2, CCSD(T)) and the hybrid density functional levels with an array of basis sets (6-31G*, 6-31G**, cc-pVDZ, 6-311+G**) were performed on 15 (CH)5SiH structures. Of the 15 structures considered on the (CH)5SiH potential energy surface, 12 structures were characterized as local minima. Two new valence isomeric forms of silabenzene, V1am and V1bm, have been identified, which lie only about 20 kcal/mol higher than silabenzene (B1). PY, where Si is hexacoordinated, has been characterized as a stationary point, which lies only around 56 kcal/mol higher in energy than silabenzene. The relative energies are contrasted with those of the valence isomers of benzene. The lower magnitudes of the vibrational frequencies corresponding to the skeletal movements support the facile rearrangements witnessed in these compounds. The chemical hardness values were measured, and no direct correlation was obtained between the relative energy ordering and the chemical hardness values.
Keep Reading...A Theoretical Study of the Structures, Energetics, Stabilities, Reactivities, and Out-of-Plane Distortive Tendencies of Skeletally Substituted Benzenes (CH)5XH and (CH)4(XH)2 (X = B-, N+, Al-, Si, P+, Ga-, Ge, and As+)
UD Priyakumar, GN Sastry,
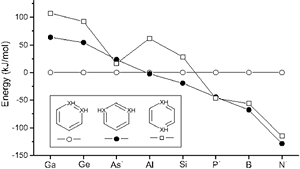
Ab initio molecular orbital theory at Hartree−Fock (HF), post-Hartree−Fock (MP2 and CCSD(T)), and the hybrid density functional theory (B3LYP) calculations were done on the mono-(CH)5XH and diskeletally substituted (CH)4(XH)2 benzenes (X = B-, N+, Al-, Si, P+, Ga-, Ge, and As+). The computed relative energies of the disubstituted isomers show interesting trends. While the ortho-isomer is the most stable for X = Ga-, Ge, and As+, meta was found to be the most stable for X = B-, N+, Al- and Si, and para was found to be the most stable for X = P+. Various intricate factors that govern the relative stabilities, such as the sum of bond strengths in the twin Kekule forms, rule of topological charge stabilization (TCS), and electrostatic repulsion were critically examined. The sum of bond strengths in the twin Kekule forms was proved to be quite a successful measure in predicting the relative stability orders between ortho- and meta-/para-isomers. The rule of TCS breaks down especially in the presence of overwhelming factors such as the differences in the cumulative bond strengths of the two positional isomers; however, the stability ordering between the para- and meta-isomers is successfully predicted in most cases. The tendency for ring puckering increases a great deal especially when the substituents are from 3rd or 4th row....
Keep Reading...Ring closure synthetic strategies toward buckybowls: benzannulation versus cyclopentannulation
UD Priyakumar, GN Sastry,
Computations were performed on idealized retrosynthetic routes towards two model buckybowl compunds, sumanene (I) and pinakene (II). Two possible paths for sumanene (I) and six for pinakene (II) were analyzed. The computational results unequivocally predict that benzannulation is a significantly easier process compared to cyclopentannulation in the ring closure strategies in both cases. The suitability of the theoretical models for obtaining reliable trends is assessed and generalizations for the synthetic strategies directed towards buckybowls and C60 were made.
Keep Reading...A computational study of the valence isomers of benzene and their group V hetero analogs
UD Priyakumar, TC Dinadayalane, GN Sastry,
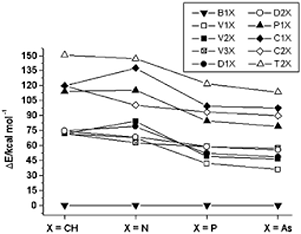
Ab initio [HF, MP2 and up to CCSD(T)] and hybrid density functional B3LYP calculations were performed on benzene, group V heterobenzenes, (CH)5X (X = N, P and As), and their corresponding valence isomers. Six valence isomers of benzene and ten valence isomers of (CH)5X have been identified and their structures, energetics, relative stabilities and their reactivities were studied. The stability ordering observed in phosphinine and arsabenzene isomers is very similar to that in the benzene isomers (benzene > benzvalene > Dewar benzene > prismane > bicyclopropenyl > trans-Dewar benzene). In the pyridine isomers, one of the bicyclopropenyl isomers (C2N) is computed to be more stable than the prismane isomer (P1N). The framework is the main factor determining the relative stabilities and the nature of the substituent is of secondary importance. Exactly the same trend in the relative energies is observed for phosphinine and arsabenzene isomers.
Keep Reading...The bicyclo [2.1. 1] hexan-2-one system: a new probe for the experimental and computational study of electronic effects in π-facial selectivity in nucleophilic additions
G Mehta, SR Singh, V Gagliardini, UD Priyakumar, GN Sastry,
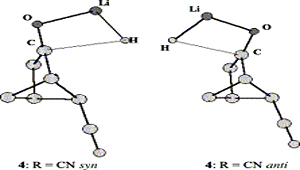
The remotely substituted 5-exo-bicyclo[2.1.1]hexan-2-one system is introduced as a new probe to study long range electronic effects on π-face selectivity during hydride reduction and a systematic computational study demonstrates good predictability at the semi-empirical level.
Keep Reading...Heterobuckybowls: a theoretical study on the structure, bowl-to-bowl inversion barrier, bond length alternation, structure-inversion barrier relationship, stability, and synthetic feasibility
UD Priyakumar, GN Sastry,
Hybrid density functional theory (DFT) calculations at the B3LYP/cc-pVDZ level have been performed on a series of heterobuckybowls, 3X, C18X3H6 (X = O, NH, CH2, BH, S, PH, PH3, Si, SiH2, and AlH). The minimum energy conformations and the transition states for bowl-to-bowl inversion, where the geometry is bowl shaped, are computed and characterized by frequency calculations. The geometries of heterotrindenes, 2X, C12X3H6 (X = O, NH, CH2, BH, S, PH, PH3, Si, SiH2, and AlH), were obtained, and the bond length alternation (Δ) in the central benzenoid ring shows remarkable sensitivity as a function of substituent with a wide range of fluctuations (−0.014 to +0.092 Å). The Δ computed in 2BH was found to be comparable with the highest bond alternation reported to date in benzenoid frameworks. The inversion dynamics of these heterobowls and their bowl depths were fit to a mixed quartic/quadratic function. The size of the heteroatom seems to exclusively control the bowl depth and rigidity as well as the synthetic feasibility. In contrast, the bond length alternation seems to be controlled by electronic factors and not by the size of the substituted atom either in trindenes or in heterosumanenes. The thermodynamic stability of this class of compounds is very much comparable with trithiasumanene (3S), which has been...
Keep Reading...Theoretical studies on the effect of sequential benzannulation to corannulene
TC Dinadayalane, UD Priyakumar, GN Sastry,
Exploratory semi-empirical SCF calculations are done on mono, di, tri, tetra, and pentabenzocorannulenes to assess the effect of sequential benzannulation to the corannulene moiety. Computed results predict a gradual reduction in the bowl-to-bowl inversion barrier in each step of the successive benzannulations and eventually for pentabenzocorannulene the barrier is only 1.4 kcal mol−1 at the MNDO level. This is also followed by gradual flattening of the central corannulene skeleton and the strain energy in benzannulated corannulenes is found to be more due to the steric repulsions between the peri-hydrogens due to the tessellation of six-membered rings. Molecular mechanics calculations are performed to evaluate the strain energy of the various benzocorannulenes. The strain energy build-up in a sequential bridging starting from pentabenzocorannulene en route to C40H10 are done. AM1 consistently overestimates the inversion barrier and based on the previous related calculations MNDO is adjudged to be the better choice to employ on this class of compounds. The sequential strain energy build-up is also evaluated starting from the pentabenzocorannulene, C40H20, to a very deep bowl, C40H10, which is the main structural motif of C60.
Keep Reading...First ab initio and density functional study on the structure, bowl-to-bowl inversion barrier, and vibrational spectra of the elusive C 3 v-Symmetric Buckybowl: Sumanene, C21H12
UD Priyakumar, GN Sastry,
The synthetically elusive C3v symmetric sumanene (C21H12), a key structural motif of C60, was subjected to a detailed computational study, exploring the structure, bowl-to-bowl inversion dynamics, vibrational spectra, and some other physicochemical properties. Hartree−Fock (HF), pure (BLYP, BP86, and BPW91), and hybrid density functional (B3LYP, B3P86, and B3PW91) calculations were done with an array of basis sets (STO-3G, 3-21G, 6-31G*, 6-31G**, 6-311G*, 6-311G**, 6-311+G*, 6-311++G*, cc-pVDZ, and cc-pVTZ). The effect of a basis set higher than double-ζ quality and the inclusion of dynamic correlation on the geometry and bowl-to-bowl inversion barrier was insignificant. The B3LYP or HF method with the cc-pVDZ or 6-311G* basis set gave satisfactory results. The previously computed modified neglect of diatomic overlap (MNDO) value of 24.2 kcal/mol for the bowl-to-bowl inversion was found to be too high, and a revised value of 16.9 kcal/mol was obtained by the B3LYP/cc-pVTZ//B3LYP/cc-pVDZ method. Consequently, the computed results indicate that sumanene (2) is not locked in the bowl geometry and that a definitive bowl-to-bowl inversion should exist at room temperature. The highest level of theory used in the study (B3LYP/6-311G**) yields values of 1.14 Å, 2.45 D, and 98.8° for the bowl depth, dipole moment, and π-orbital axis vector angle at the hub carbon for sumanene, respectively....
Keep Reading...An ab initio and DFT study of the valence isomers of pyridine
UD Priyakumar, TC Dinadayalane, GN Sastry,
Ab initio (HF, MP2, and CCSD(T)) and density functional theory computed results on the equilibrium geometries, relative stabilities, strain energies, and vibrational spectra of the nine possible valence isomers of pyridine are reported. Although some aza-benzvalenes (V1N and V3N) lie lower in energy than Dewar pyridines (D1N and D2N), the strain energies for the latter are lower. Relative stabilities of the valence isomers, thermodynamic stability, and skeletal rigidity are comparable to those of benzene valence isomers.
Keep Reading...Tailoring the curvature, bowl rigidity and stability of heterobuckybowls: theoretical design of synthetic strategies towards heterosumanenes
UD Priyakumar, GN Sastry,
Quantum mechanical calculations predict that larger heteroatom substituents on the periphery increase the feasibility of the crucial third ring closure in sumanene and are responsible for the accompanying modulations in the curvature, rigidity, stability and some of the physicochemical properties of the resulting heterosumanenes. Systematic application of semiempirical, ab initio, and DFT methods reveal that the qualitative trends obtained and our principal conclusions are independent of level of theory, albeit with minor quantitative differences.
Keep Reading...Structures, energetics and vibrational spectra of the valence isomers of phosphinine. An ab initio and DFT study,
UD Priyakumar, TC Dinadayalane, GN Sastry,

The structures and energetics of nine valence isomers of phosphinine, (CH)5P, have been investigated by ab initio (HF, MP2 and CCSD(T)) and hybrid density functional (B3LYP) methods. The relative stability ordering of the (CH)5P isomers is similar to those of (CH)6. Strain energies are evaluated for all the non-planar isomers based on the sum of standard bond strengths, taking the planar resonance stabilized isomer, phosphinine as the reference. Lower magnitudes of the frequencies corresponding to the first few normal modes compared to their benzene isomers account for smoother isomerization reactions among them.
Keep Reading...Theory provides a clue to accomplish the synthesis of sumanene, C21H12, the prototypical C3v-buckybowl,
UD Priyakumar, GN Sastry,
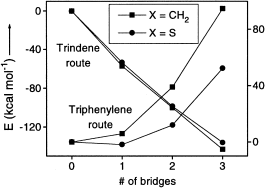
Two possible synthetic routes, namely a triphenylene route and a trindene route for the synthesis of sumanene, C21H12 (2CH2) and trithiasumanene, C18S3H6 (2S) are analyzed in detail. Theoretical calculations unequivocally predict that the synthesis of sumanene may be easily achieved through a trindene route. Here is a clue to achieve the synthesis of sumanene. Theoretical calculations predict that the synthesis of sumanene can be achieved if one starts from appropriately substituted trindene.
Keep Reading...The role of heteroatom substitution in the rigidity and curvature of buckybowls. A theoretical study,
GN Sastry, UD Priyakumar,
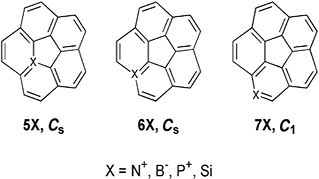
Effect of substitution on the curvature and bowl-to-bowl inversion barrier of bucky-bowls. Study of mono-substituted corannulenes (C19XH10, X= B−, N+, P+ and Si),
GN Sastry, HSP Rao, P Bednarek, UD Priyakumar,
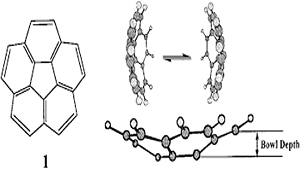
Structure, stability and reactivity parameters of (CH)8 isomers and their cation and anion radical counterparts : A theoretical study,
UD Priyakumar, GN Sastry,
Among the hydrocarbons with (CH)2k (k = 1,2,3,4 .... ) structural formula. (CH)8 where k=4, possesses nineteen local minima at semiempirical level with comparable heats of formation. AM1 procedure is found to be better than MNDO and PM3 in evaluating the heats of formation and geometries based on comparison with available experimental data of the neutral isomers. Vertical and adiabatic electron affinities and ionization potentials have been calculated for all the available isomers. The relative energy ordering is mainly controlled by the electronic factors rather than the strain. Koopman's theorem is not expected to yield reliable answers for ionization potentials, as the orbital relaxation seems to be very high for some isomers. The major geometric distortions are due to either Jahn-Teller distortions or strong vibronic interactions and the consequent reordering of the skeleton. Relaxation energies from vertical to adiabatic states are roughly proportional to the geometric deformations that occur upon ionization or addition of an electron.
Keep Reading...Structures, Energetics, Relative Stabilities, and Out-of-Plane Distortivities of Skeletally Disubstituted Benzenes, (CH) 4X2 (X= N, P, C-, Si-, O+, and S+): An ab Initio and DFT study,
UD Priyakumar, GN Sastry,
The positional isomers of disubstituted benzenes, (CH)4X2 (X = N, P, C-, Si-, O+, and S+), are studied using ab initio molecular orbital theory at Hartree−Fock (HF), MP2, and CCSD(T) levels and also using density functional theory (B3LYP). All of the planar structures are characterized as minima, and they show fully delocalized geometric parameters, with no significant sign of bond fixation. The ortho isomer is computed to be the least stable in all cases except when X = P and Si-. While meta isomer is more stable than para in general, exceptions are seen for C- and S+ substitutions. Various factors that affect the relative stabilities of positional isomers, viz., (a) lone pair−lone pair (lp−lp) repulsion, (b) electrostatic interaction, (c) bond strengths in the corresponding valence isomers, and (d) topological charge stabilization (TCS), are invoked to explain the computed pattern of relative stabilities. The strengths and limitations of each of these aspects in controlling the geometries and energetics of the three isomeric forms are assessed. The lp−lp interaction is found to be insignificant in the isomers bearing third row atom substituents. The rule of TCS excellently accounts for the distinction between meta and para isomers. The sum of the bond strengths of the constituent Kekule forms derived from...
Keep Reading...